Department of Ecology, Evolution, and Natural Resources, School of Environmental and Biological Sciences Rutgers, The State University of New Jersey, 14 College Farm Rd, New Brunswick, NJ 08901, USA.
BMC Evol Biol. 2012 May 30;12:74. doi: 10.1186/1471-2148-12-74.
The literature is ripe with phylogenetic estimates of nucleotide substitution rates, especially of measurably evolving species such as RNA viruses. However, it is not known how robust these rate estimates are to inaccuracies in the data, particularly in sampling dates that are used for molecular clock calibration. Here we report on the rate of evolution of the emerging pathogen Rabbit hemorrhagic disease virus (RHDV), which has significantly different rates of evolution for the same outer capsid (VP60) gene published in the literature. In an attempt to reconcile the conflicting data and further elucidate details of RHDV 's evolutionary history, we undertook fresh Bayesian analyses and employed jackknife control methods to produce robust substitution rate and time to most recent common ancestor (TMRCA) estimates for RHDV based on the VP60 and RNA-dependent RNA polymerase genes.
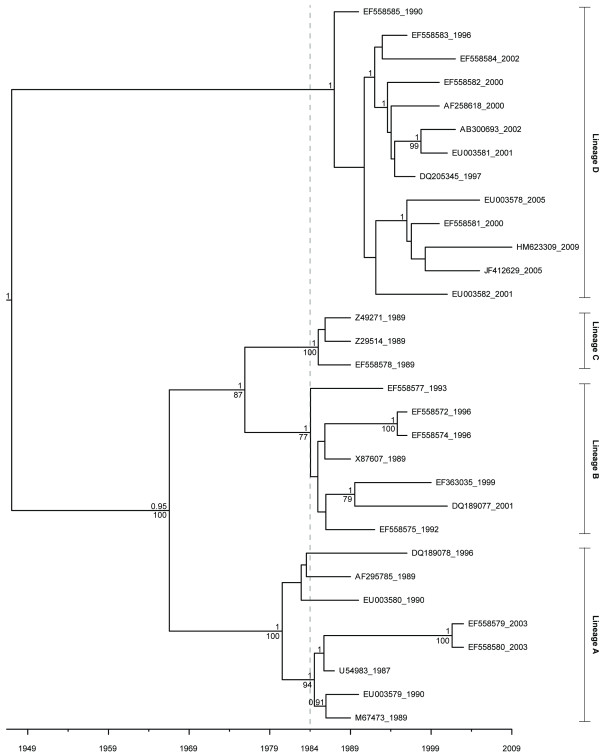
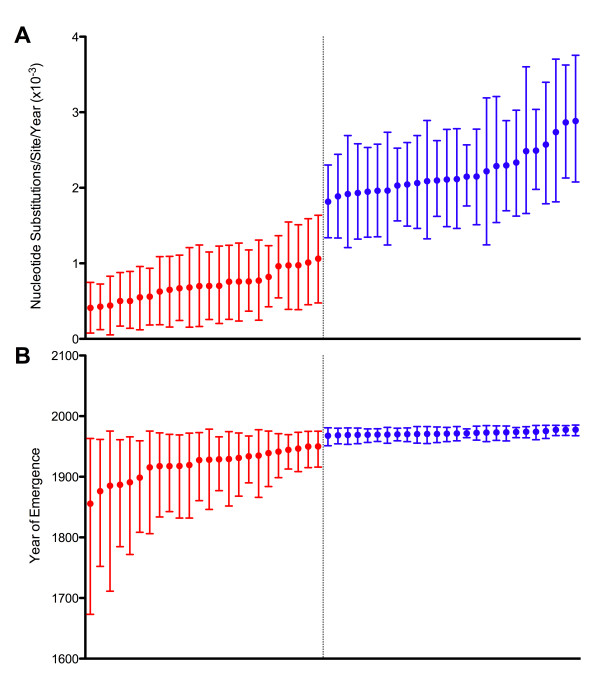
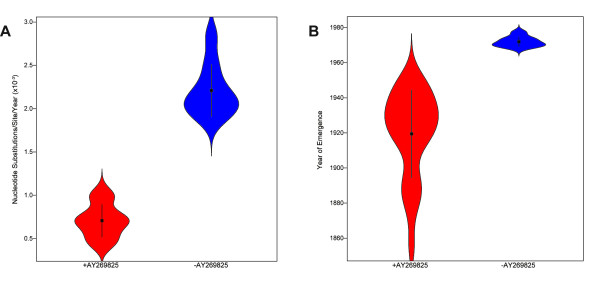
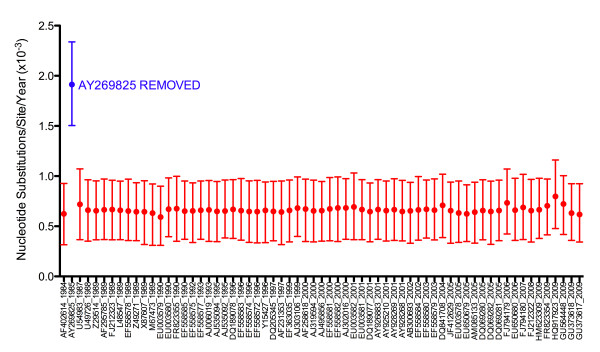
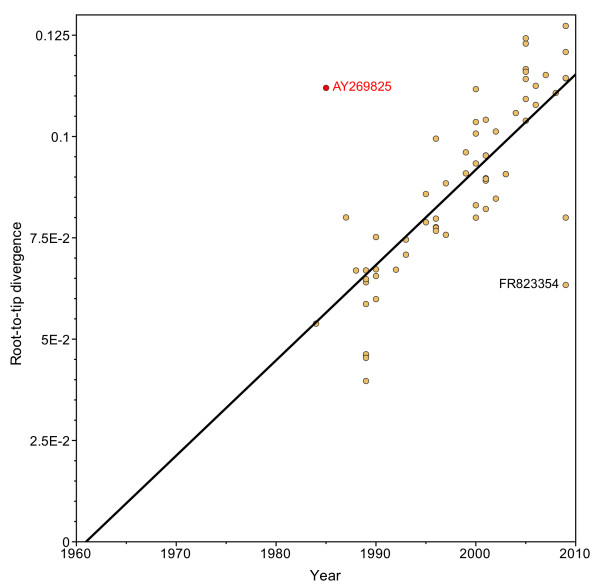
Through these control methods, we were able to identify a single misdated taxon, a passaged lab strain used for vaccine production, which was responsible for depressing the RHDV capsid gene's rate of evolution by 65%. Without this isolate, the polymerase and the capsid protein genes had nearly identical rates of evolution: 1.90x10-3 nucleotide substitutions/site/year, ns/s/y, (95% highest probability density (HPD) 1.25x10-3-2.55x10-3) and 1.91x10-3 ns/s/y (95% HPD 1.50x10-3-2.34x10-3), respectively.
After excluding the misdated taxon, both genes support a significantly higher substitution rate as well as a relatively recent emergence of RHDV, and obviate the need for previously hypothesized decades of unobserved diversification of the virus. The control methods show that using even one misdated taxon in a large dataset can significantly skew estimates of evolutionary parameters and suggest that it is better practice to use smaller datasets composed of taxa with unequivocal isolation dates. These jackknife controls would be useful for future tip-calibrated rate analyses that include taxa with ambiguous dates of isolation.
文献中充满了核苷酸替代率的系统发育估计值,特别是对于可测量进化的物种,如 RNA 病毒。然而,目前尚不清楚这些速率估计值对数据的不准确程度有多稳健,特别是在用于分子钟校准的采样日期。在这里,我们报告了新兴病原体兔出血症病毒 (RHDV) 的进化速度,文献中公布的相同外壳 (VP60) 基因的进化速度有显著差异。为了协调相互矛盾的数据,并进一步阐明 RHDV 进化历史的细节,我们进行了新的贝叶斯分析,并采用了自举控制方法,根据 VP60 和 RNA 依赖性 RNA 聚合酶基因,为 RHDV 产生稳健的替代率和最近共同祖先 (TMRCA) 估计值。
通过这些控制方法,我们能够识别出一个单独的错误日期分类单元,这是一种用于疫苗生产的传代实验室菌株,它使 RHDV 外壳蛋白基因的进化速度降低了 65%。如果没有这个分离株,聚合酶和外壳蛋白基因的进化速度几乎相同:1.90x10-3 个核苷酸取代/位点/年,ns/s/y(95%最高概率密度 (HPD) 1.25x10-3-2.55x10-3)和 1.91x10-3 ns/s/y(95% HPD 1.50x10-3-2.34x10-3)。
排除错误日期的分类单元后,两个基因都支持更高的替代率以及 RHDV 的相对近期出现,并消除了病毒在数十年未观察到的多样化的先前假设。控制方法表明,即使在大型数据集中有一个错误日期的分类单元,也会显著扭曲进化参数的估计值,并表明使用具有明确隔离日期的小数据集组成更可取。这些自举控制方法将对未来包括隔离日期不明确的分类单元的尖峰校准速率分析有用。