Rare Brain Disorders Clinic and Laboratory, Department of Neurology and Neurotherapeutics, UT Southwestern Medical Center, USA.
Neurobiol Dis. 2012 Oct;48(1):92-101. doi: 10.1016/j.nbd.2012.04.011. Epub 2012 Apr 23.
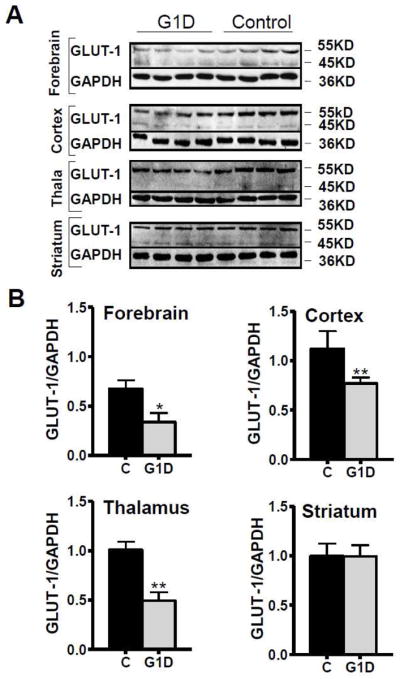
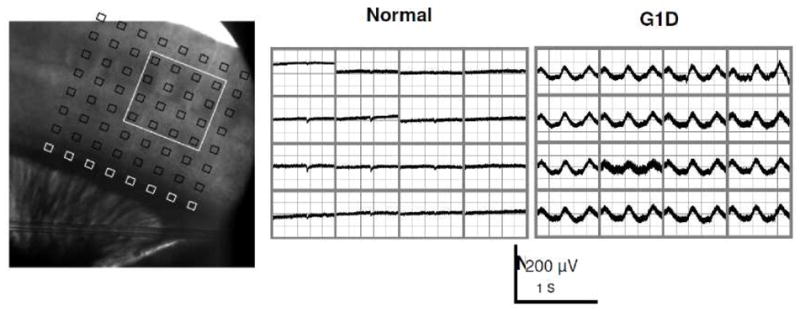
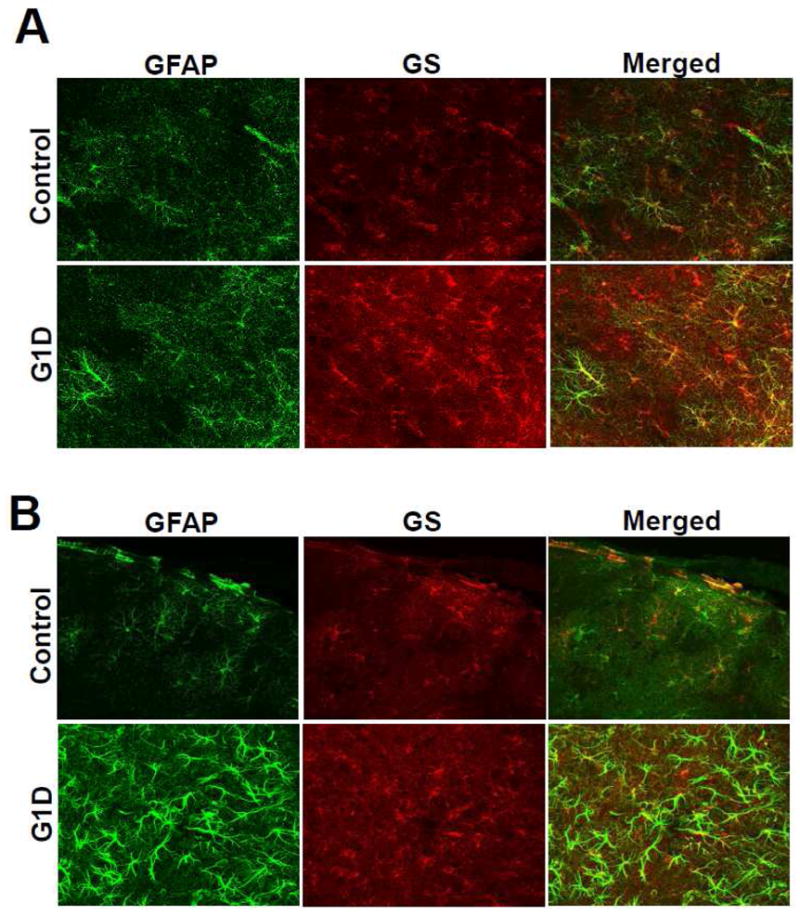
Brain glucose supplies most of the carbon required for acetyl-coenzyme A (acetyl-CoA) generation (an important step for myelin synthesis) and for neurotransmitter production via further metabolism of acetyl-CoA in the tricarboxylic acid (TCA) cycle. However, it is not known whether reduced brain glucose transporter type I (GLUT-1) activity, the hallmark of the GLUT-1 deficiency (G1D) syndrome, leads to acetyl-CoA, TCA or neurotransmitter depletion. This question is relevant because, in its most common form in man, G1D is associated with cerebral hypomyelination (manifested as microcephaly) and epilepsy, suggestive of acetyl-CoA depletion and neurotransmitter dysfunction, respectively. Yet, brain metabolism in G1D remains underexplored both theoretically and experimentally, partly because computational models of limited brain glucose transport are subordinate to metabolic assumptions and partly because current hemizygous G1D mouse models manifest a mild phenotype not easily amenable to investigation. In contrast, adult antisense G1D mice replicate the human phenotype of spontaneous epilepsy associated with robust thalamocortical electrical oscillations. Additionally, and in consonance with human metabolic imaging observations, thalamus and cerebral cortex display the lowest GLUT-1 expression and glucose uptake in the mutant mouse. This depletion of brain glucose is associated with diminished plasma fatty acids and elevated ketone body levels, and with decreased brain acetyl-CoA and fatty acid contents, consistent with brain ketone body consumption and with stimulation of brain beta-oxidation and/or diminished cerebral lipid synthesis. In contrast with other epilepsies, astrocyte glutamine synthetase expression, cerebral TCA cycle intermediates, amino acid and amine neurotransmitter contents are also intact in G1D. The data suggest that the TCA cycle is preserved in G1D because reduced glycolysis and acetyl-CoA formation can be balanced by enhanced ketone body utilization. These results are incompatible with global cerebral energy failure or with neurotransmitter depletion as responsible for epilepsy in G1D and point to an unknown mechanism by which glycolysis critically regulates cortical excitability.
大脑葡萄糖提供了生成乙酰辅酶 A(乙酰-CoA)(髓鞘合成的重要步骤)和通过三羧酸 (TCA) 循环中乙酰-CoA 的进一步代谢产生神经递质所需的大部分碳。然而,尚不清楚减少脑葡萄糖转运蛋白 1 (GLUT-1) 活性(GLUT-1 缺乏症 (G1D) 综合征的标志)是否会导致乙酰-CoA、TCA 或神经递质耗竭。这个问题很重要,因为在其在人类中最常见的形式中,G1D 与脑发育不全(表现为小头症)和癫痫有关,分别提示乙酰-CoA 耗竭和神经递质功能障碍。然而,G1D 的脑代谢在理论和实验上都尚未得到充分探索,部分原因是脑葡萄糖转运的计算模型受制于代谢假设,部分原因是当前的半合子 G1D 小鼠模型表现出轻度表型,不易进行研究。相比之下,成年反义 G1D 小鼠复制了与强大的丘脑皮质电振荡相关的自发性癫痫的人类表型。此外,与人类代谢成像观察结果一致,在突变小鼠中,丘脑和大脑皮层显示出最低的 GLUT-1 表达和葡萄糖摄取。这种脑葡萄糖的耗竭与血浆脂肪酸减少和酮体水平升高有关,并且与脑乙酰-CoA 和脂肪酸含量减少有关,这与脑酮体消耗以及脑β-氧化刺激和/或脑脂质合成减少有关。与其他癫痫不同,G1D 中的星形胶质细胞谷氨酰胺合酶表达、脑 TCA 循环中间产物、氨基酸和胺神经递质含量也完整无缺。这些数据表明,TCA 循环在 G1D 中得以保留,因为减少的糖酵解和乙酰-CoA 形成可以通过增强酮体利用来平衡。这些结果与脑能量衰竭或神经递质耗竭导致 G1D 中的癫痫发作不一致,并指向一个未知的机制,即糖酵解对皮质兴奋性具有关键调节作用。