Buck Institute for Research on Aging, 8001 Redwood Blvd., Novato, CA 94945, USA.
Integr Biol (Camb). 2012 Jul;4(7):795-804. doi: 10.1039/c2ib00136e. Epub 2012 Jun 18.
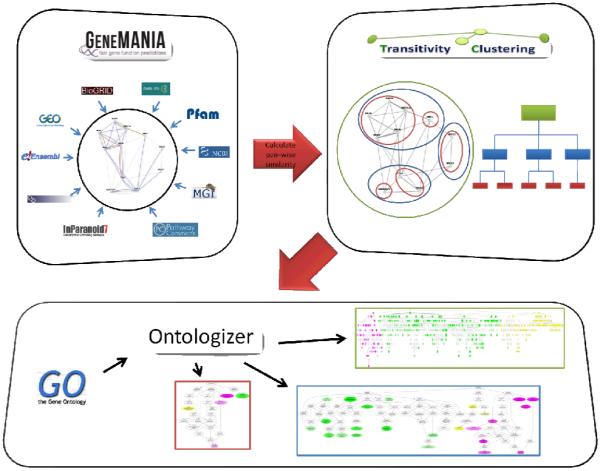
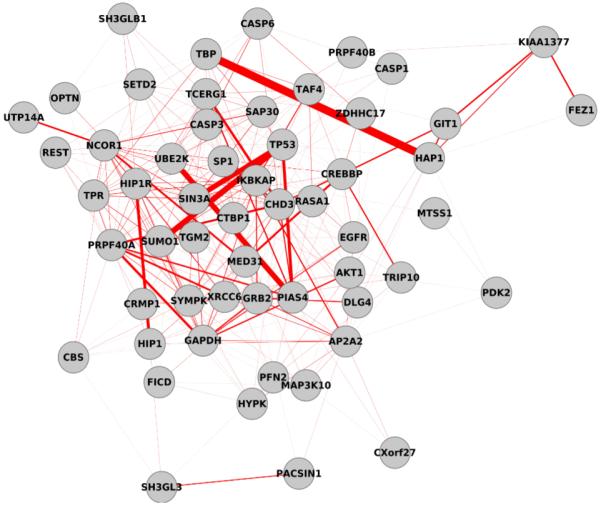
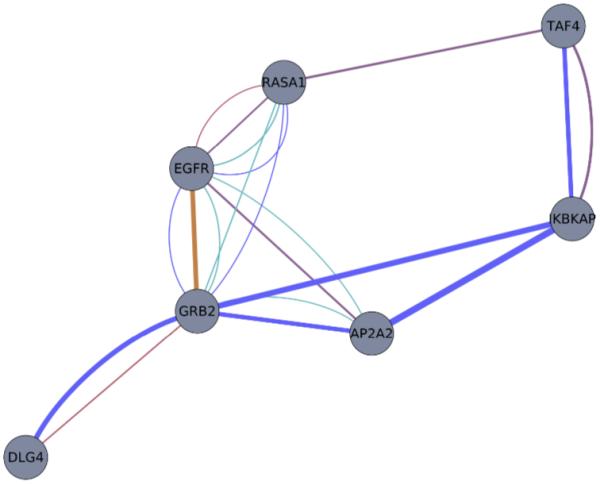
High-throughput biological experiments commonly result in a list of genes or proteins of interest. In order to understand the observed changes of the genes and to generate new hypotheses, one needs to understand the functions and roles of the genes and how those functions relate to the experimental conditions. Typically, statistical tests are performed in order to detect enriched Gene Ontology categories or pathways, i.e. the categories are observed in the genes of interest more often than is expected by chance. Depending on the number of genes and the complexity and quantity of functions in which they are involved, such an analysis can easily result in hundreds of enriched terms. To this end we developed DEFOG, a web-based application that facilitates the functional analysis of gene sets by hierarchically organizing the genes into functionally related modules. Our computational pipeline utilizes three powerful tools to achieve this goal: (1) GeneMANIA creates a functional consensus network of the genes of interest based on gene-list-specific data fusion of hundreds of genomic networks from publicly available sources; (2) Transitivity Clustering organizes those genes into a clear hierarchy of functionally related groups, and (3) Ontologizer performs a Gene Ontology enrichment analysis on the resulting gene clusters. DEFOG integrates this computational pipeline within an easy-to-use web interface, thus allowing for a novel visual analysis of gene sets that aids in the discovery of potentially important biological mechanisms and facilitates the creation of new hypotheses. DEFOG is available at http://www.mooneygroup.org/defog.
高通量生物实验通常会产生一组感兴趣的基因或蛋白质。为了理解观察到的基因变化并生成新的假设,需要了解基因的功能和作用,以及这些功能如何与实验条件相关。通常,会进行统计检验,以检测基因本体论 (GO) 类别或途径的富集,即观察到的基因本体论类别在感兴趣的基因中比随机出现的频率更高。根据基因的数量以及它们所涉及的功能的复杂性和数量,这种分析很容易导致数百个富集术语。为此,我们开发了 DEFOG,这是一种基于网络的应用程序,通过将基因分层组织到功能相关的模块中,为基因集的功能分析提供便利。我们的计算流程利用三个强大的工具来实现这一目标:(1)GeneMANIA 根据来自公开来源的数百个基因组网络的基因列表特定数据融合,为感兴趣的基因创建功能共识网络;(2)传递聚类将这些基因组织成功能相关的明确层次结构组;(3)Ontologizer 对生成的基因簇进行基因本体论富集分析。DEFOG 将这个计算流程集成到一个易于使用的网络界面中,从而实现了基因集的新的可视化分析,有助于发现潜在的重要生物学机制并促进新假设的创建。DEFOG 可在 http://www.mooneygroup.org/defog 上获得。