Department of Biosciences, University of Eastern Finland, PO Box 1627, FIN-70211 Kuopio, Finland.
BMC Bioinformatics. 2011 May 19;12:171. doi: 10.1186/1471-2105-12-171.
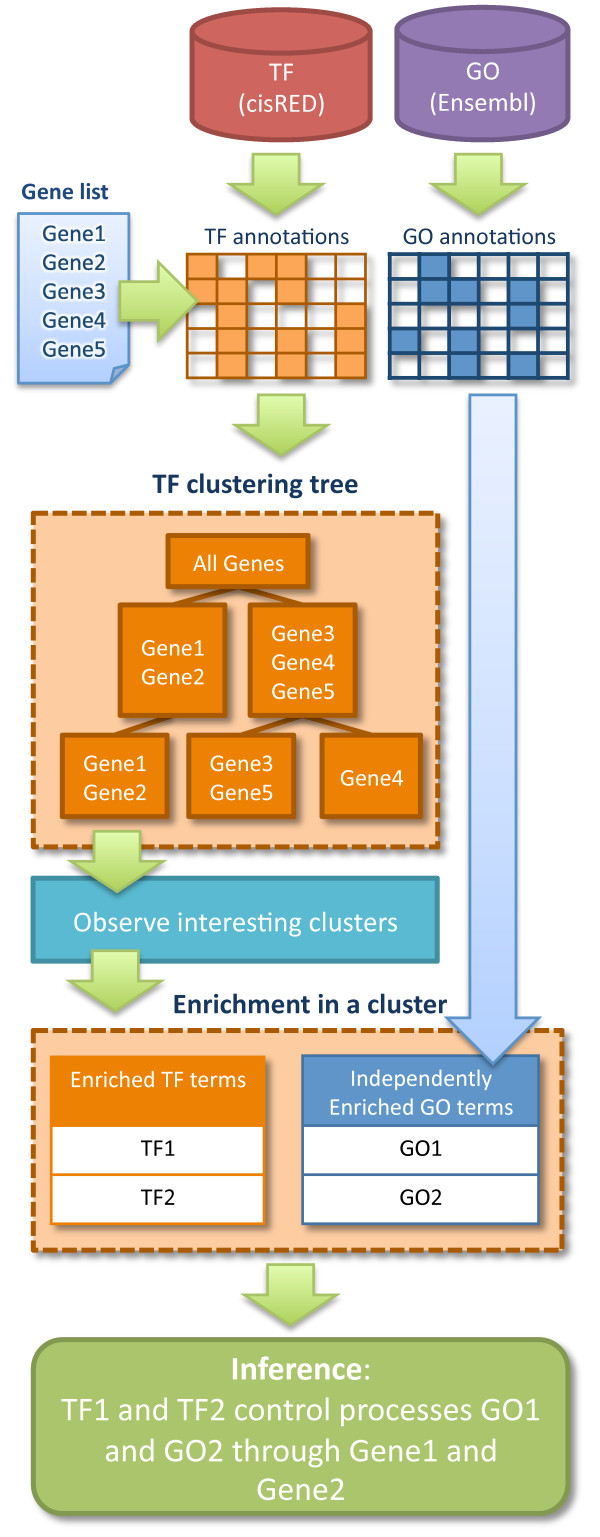
A major challenge in genomic research is identifying significant biological processes and generating new hypotheses from large gene sets. Gene sets often consist of multiple separate biological pathways, controlled by distinct regulatory mechanisms. Many of these pathways and the associated regulatory mechanisms might be obscured by a large number of other significant processes and thus not identified as significant by standard gene set enrichment analysis tools.
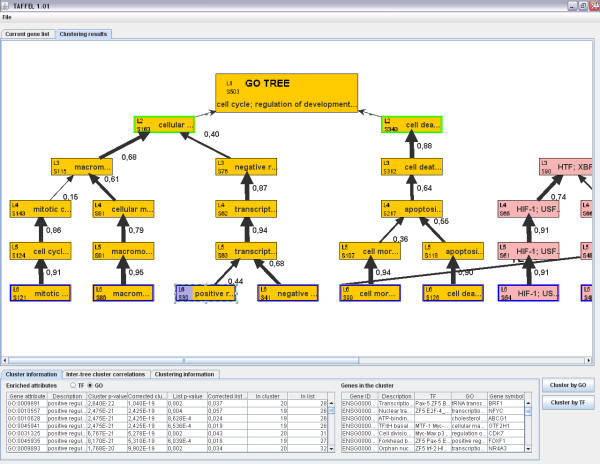
We present a novel method called Independent Enrichment Analysis (IEA) and software TAFFEL that eases the task by clustering genes to subgroups using Gene Ontology categories and transcription regulators. IEA indicates transcriptional regulators putatively controlling biological functions in studied condition.
We demonstrate that the developed method and TAFFEL tool give new insight to the analysis of differentially expressed genes and can generate novel hypotheses. Our comparison to other popular methods showed that the IEA method implemented in TAFFEL can find important biological phenomena, which are not reported by other methods.
基因组研究中的一个主要挑战是从大型基因集中识别重要的生物过程并生成新的假设。基因集通常由多个独立的生物途径组成,由不同的调控机制控制。其中许多途径和相关的调控机制可能会被大量其他重要过程所掩盖,因此不会被标准的基因集富集分析工具识别为重要。
我们提出了一种新的方法,称为独立富集分析(IEA)和软件 TAFFEL,它通过使用基因本体论类别和转录调节剂将基因聚类到子组中,从而简化了任务。IEA 指示在研究条件下可能控制生物功能的转录调节剂。
我们证明,所开发的方法和 TAFFEL 工具为差异表达基因的分析提供了新的见解,并可以生成新的假设。我们与其他流行方法的比较表明,TAFFEL 中实现的 IEA 方法可以发现其他方法未报告的重要生物学现象。