Institute of Structural and Molecular Biology, University College of London, London, UK.
BMC Genomics. 2012 Jul 2;13:296. doi: 10.1186/1471-2164-13-296.
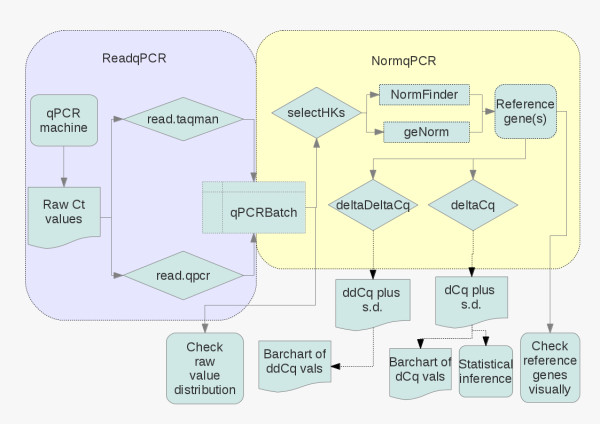
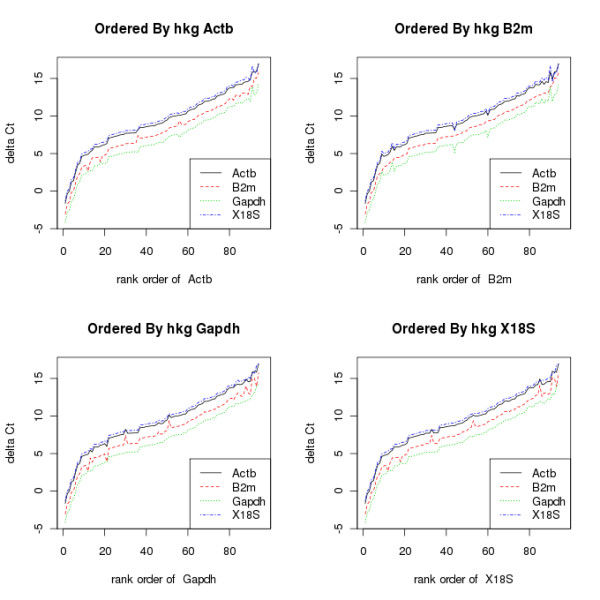
Measuring gene transcription using real-time reverse transcription polymerase chain reaction (RT-qPCR) technology is a mainstay of molecular biology. Technologies now exist to measure the abundance of many transcripts in parallel. The selection of the optimal reference gene for the normalisation of this data is a recurring problem, and several algorithms have been developed in order to solve it. So far nothing in R exists to unite these methods, together with other functions to read in and normalise the data using the chosen reference gene(s).
We have developed two R/Bioconductor packages, ReadqPCR and NormqPCR, intended for a user with some experience with high-throughput data analysis using R, who wishes to use R to analyse RT-qPCR data. We illustrate their potential use in a workflow analysing a generic RT-qPCR experiment, and apply this to a real dataset. Packages are available from http://www.bioconductor.org/packages/release/bioc/html/ReadqPCR.htmland http://www.bioconductor.org/packages/release/bioc/html/NormqPCR.html
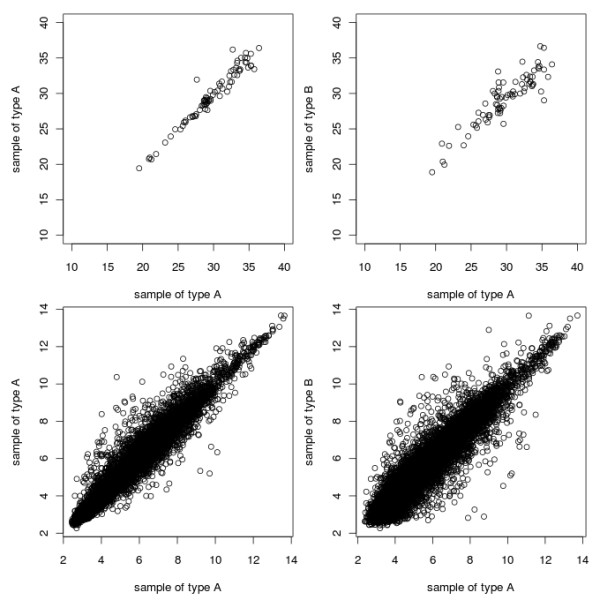
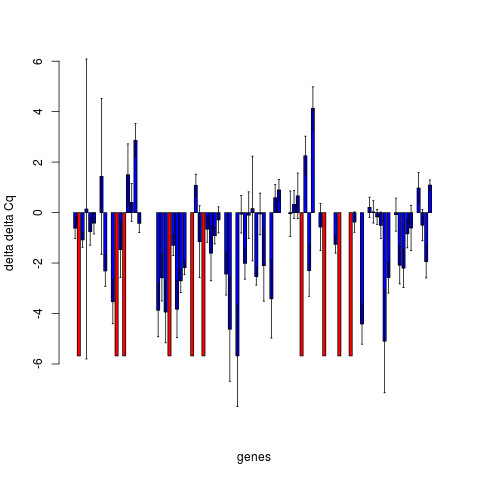
These packages increase the repetoire of RT-qPCR analysis tools available to the R user and allow them to (amongst other things) read their data into R, hold it in an ExpressionSet compatible R object, choose appropriate reference genes, normalise the data and look for differential expression between samples.
使用实时逆转录聚合酶链反应 (RT-qPCR) 技术测量基因转录是分子生物学的主要手段。现在存在同时测量许多转录本丰度的技术。选择用于规范化此数据的最佳参考基因是一个反复出现的问题,已经开发了几种算法来解决它。到目前为止,R 中还没有任何功能可以将这些方法与其他功能结合使用,以便使用所选的参考基因读取和规范化数据。
我们开发了两个 R/Bioconductor 包,ReadqPCR 和 NormqPCR,供具有使用 R 进行高通量数据分析经验的用户使用,他们希望使用 R 分析 RT-qPCR 数据。我们通过一个通用 RT-qPCR 实验的工作流程来说明它们的潜在用途,并将其应用于真实数据集。这些包可从 http://www.bioconductor.org/packages/release/bioc/html/ReadqPCR.htmland http://www.bioconductor.org/packages/release/bioc/html/NormqPCR.html 获得。
这些包增加了 R 用户可用的 RT-qPCR 分析工具的种类,并允许他们(除其他外)将数据读入 R 中,将其存储在兼容 ExpressionSet 的 R 对象中,选择适当的参考基因,规范化数据并寻找样本之间的差异表达。