Institut des Neurosciences de Montpellier, U1051 de l'INSERM, Université de Montpellier I et II, BP 74103, F-34091 Montpellier cedex 05, France.
Orphanet J Rare Dis. 2012 Jul 9;7:46. doi: 10.1186/1750-1172-7-46.
DEFINITION OF THE DISEASE: Dominant Optic Atrophy (DOA) is a neuro-ophthalmic condition characterized by a bilateral degeneration of the optic nerves, causing insidious visual loss, typically starting during the first decade of life. The disease affects primary the retinal ganglion cells (RGC) and their axons forming the optic nerve, which transfer the visual information from the photoreceptors to the lateral geniculus in the brain.
The prevalence of the disease varies from 1/10000 in Denmark due to a founder effect, to 1/30000 in the rest of the world.
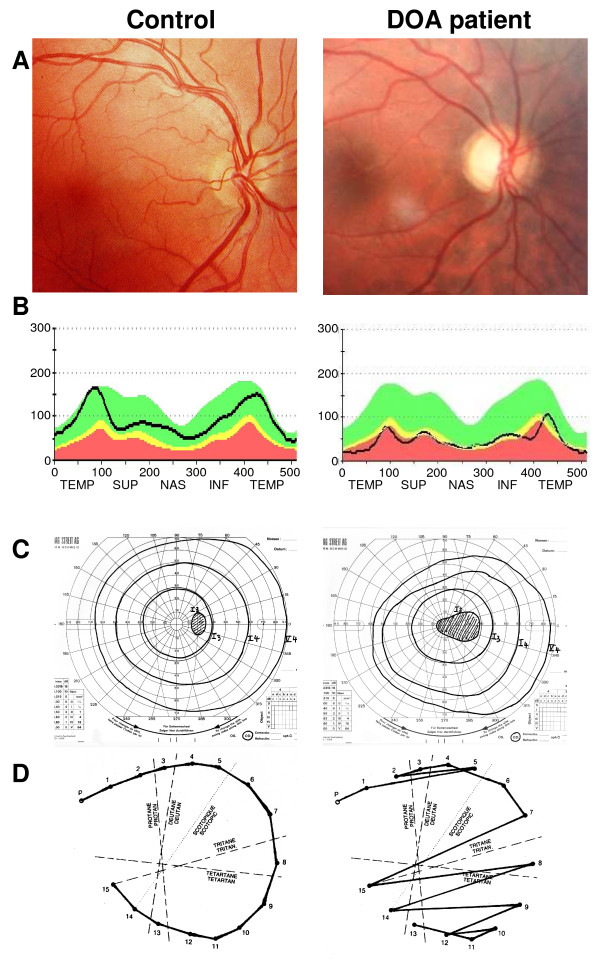
DOA patients usually suffer of moderate visual loss, associated with central or paracentral visual field deficits and color vision defects. The severity of the disease is highly variable, the visual acuity ranging from normal to legal blindness. The ophthalmic examination discloses on fundoscopy isolated optic disc pallor or atrophy, related to the RGC death. About 20% of DOA patients harbour extraocular multi-systemic features, including neurosensory hearing loss, or less commonly chronic progressive external ophthalmoplegia, myopathy, peripheral neuropathy, multiple sclerosis-like illness, spastic paraplegia or cataracts.
Two genes (OPA1, OPA3) encoding inner mitochondrial membrane proteins and three loci (OPA4, OPA5, OPA8) are currently known for DOA. Additional loci and genes (OPA2, OPA6 and OPA7) are responsible for X-linked or recessive optic atrophy. All OPA genes yet identified encode mitochondrial proteins embedded in the inner membrane and ubiquitously expressed, as are the proteins mutated in the Leber Hereditary Optic Neuropathy. OPA1 mutations affect mitochondrial fusion, energy metabolism, control of apoptosis, calcium clearance and maintenance of mitochondrial genome integrity. OPA3 mutations only affect the energy metabolism and the control of apoptosis.
Patients are usually diagnosed during their early childhood, because of bilateral, mild, otherwise unexplained visual loss related to optic discs pallor or atrophy, and typically occurring in the context of a family history of DOA. Optical Coherence Tomography further discloses non-specific thinning of retinal nerve fiber layer, but a normal morphology of the photoreceptors layers. Abnormal visual evoked potentials and pattern ERG may also reflect the dysfunction of the RGCs and their axons. Molecular diagnosis is provided by the identification of a mutation in the OPA1 gene (75% of DOA patients) or in the OPA3 gene (1% of patients).
Visual loss in DOA may progress during puberty until adulthood, with very slow subsequent chronic progression in most of the cases. On the opposite, in DOA patients with associated extra-ocular features, the visual loss may be more severe over time.
To date, there is no preventative or curative treatment in DOA; severely visually impaired patients may benefit from low vision aids. Genetic counseling is commonly offered and patients are advised to avoid alcohol and tobacco consumption, as well as the use of medications that may interfere with mitochondrial metabolism. Gene and pharmacological therapies for DOA are currently under investigation.
疾病定义:显性视神经萎缩(DOA)是一种神经眼科疾病,其特征为双侧视神经退行性变,导致隐匿性视力丧失,通常始于生命的第一个十年。该疾病主要影响视网膜神经节细胞(RGC)及其形成视神经的轴突,将视觉信息从光感受器传输到大脑中的外侧膝状体。
由于创始效应,丹麦的该病患病率为 1/10000,而世界其他地区为 1/30000。
DOA 患者通常患有中度视力丧失,伴有中心或旁中心视野缺损和色觉缺陷。疾病的严重程度差异很大,视力从正常到法定失明不等。眼科检查显示眼底孤立性视神经盘苍白或萎缩,与 RGC 死亡有关。约 20%的 DOA 患者存在眼外多系统特征,包括感觉神经性听力损失,或较少见的慢性进行性眼外肌麻痹、肌病、周围神经病、多发性硬化样疾病、痉挛性截瘫或白内障。
目前已知有两个(OPA1、OPA3)编码线粒体内膜蛋白的基因和三个(OPA4、OPA5、OPA8)位点导致 DOA。其他(OPA2、OPA6 和 OPA7)位点和基因负责 X 连锁或隐性视神经萎缩。所有已鉴定的 OPA 基因均编码嵌入在内膜中的线粒体蛋白,并广泛表达,就像在 Leber 遗传性视神经病变中突变的蛋白一样。OPA1 突变影响线粒体融合、能量代谢、细胞凋亡的控制、钙清除和线粒体基因组完整性的维持。OPA3 突变仅影响能量代谢和细胞凋亡的控制。
患者通常在其儿童早期被诊断,因为双侧、轻度、无其他原因的视力丧失与视神经盘苍白或萎缩有关,并且通常发生在 DOA 家族史的背景下。光相干断层扫描进一步显示视网膜神经纤维层的非特异性变薄,但光感受器层的形态正常。异常的视觉诱发电位和图案 ERG 也可能反映 RGC 及其轴突的功能障碍。分子诊断是通过鉴定 OPA1 基因(75%的 DOA 患者)或 OPA3 基因(1%的患者)中的突变来提供的。
DOA 患者的视力丧失可能在青春期至成年期持续进展,在大多数情况下随后的慢性进展非常缓慢。相反,在伴有眼外特征的 DOA 患者中,随着时间的推移,视力丧失可能更为严重。
迄今为止,DOA 没有预防或治疗方法;严重视力受损的患者可能受益于低视力辅助器具。通常提供遗传咨询,并建议患者避免饮酒和吸烟,以及使用可能干扰线粒体代谢的药物。DOA 的基因和药物治疗目前正在研究中。