UCL Institute of Ophthalmology, 11-43 Bath Street, London EC1V 9EL, UK.
Department of Biology, University of Padua, and Veneto Institute of Molecular Medicine, Padua 35129, Italy.
Hum Mol Genet. 2022 Oct 10;31(20):3478-3493. doi: 10.1093/hmg/ddac128.
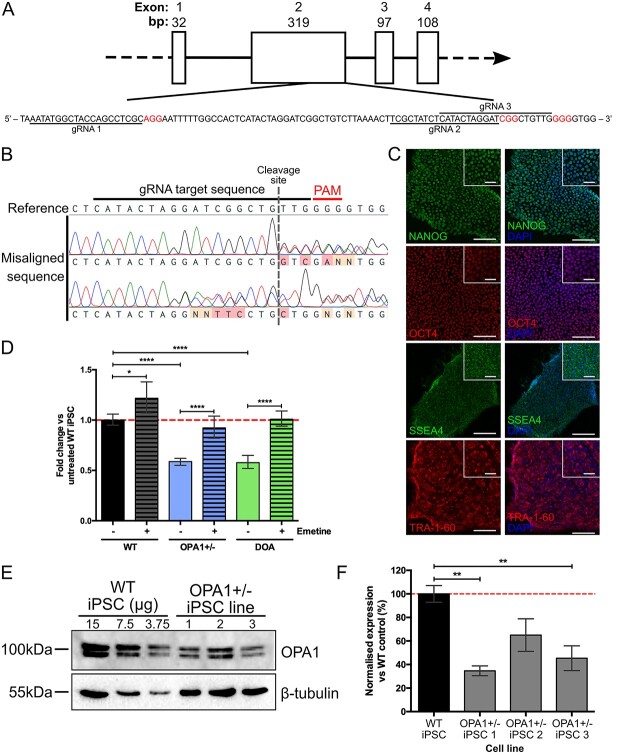
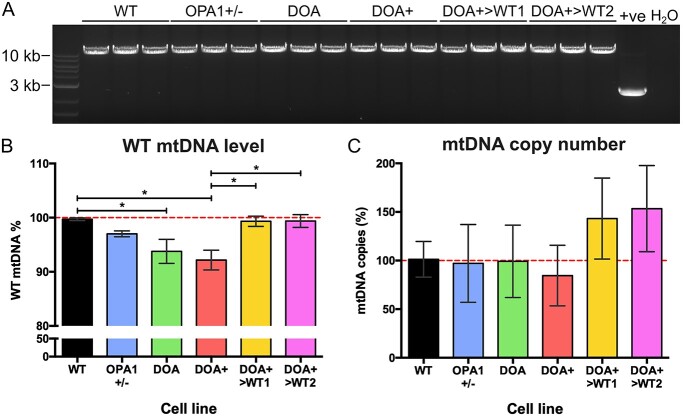
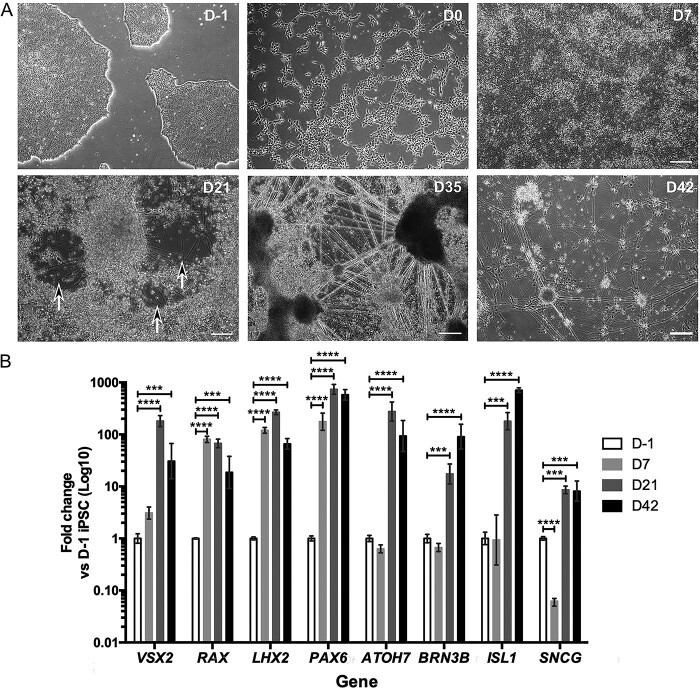
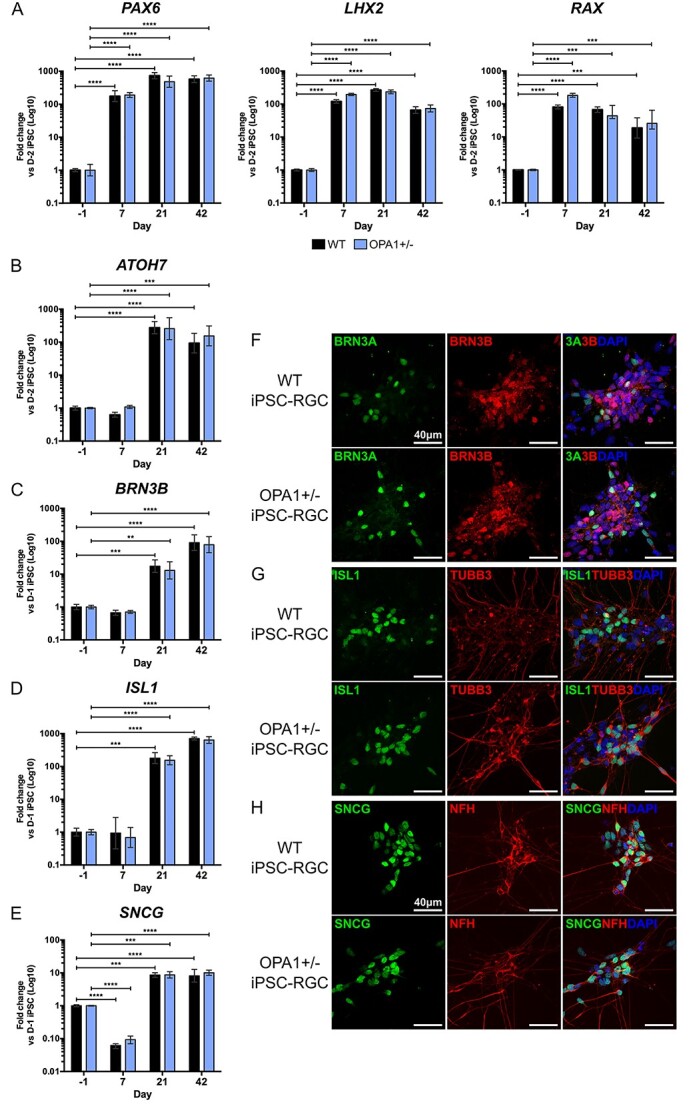
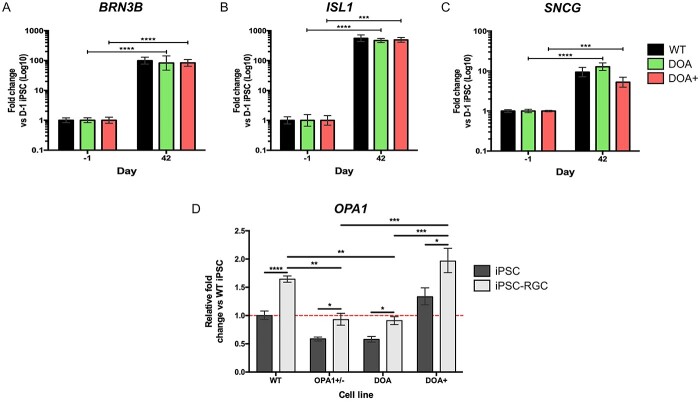
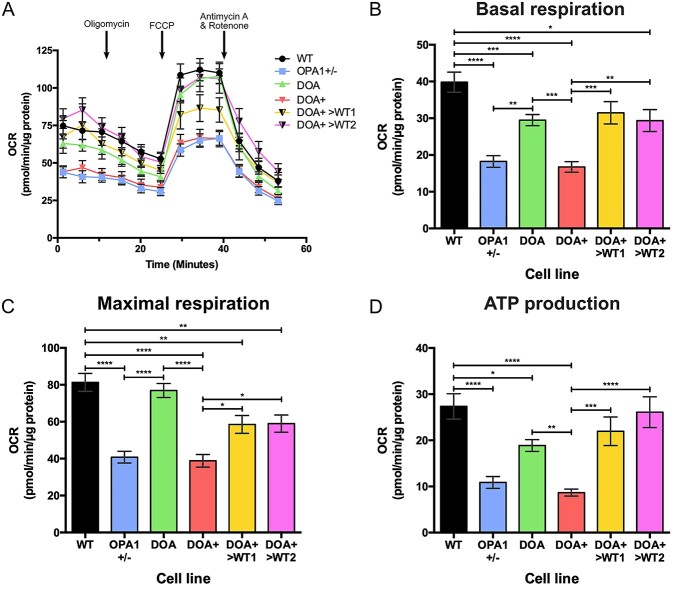
Autosomal dominant optic atrophy (DOA) is the most common inherited optic neuropathy, characterized by the preferential loss of retinal ganglion cells (RGCs), resulting in optic nerve degeneration and progressive bilateral central vision loss. More than 60% of genetically confirmed patients with DOA carry variants in the nuclear OPA1 gene, which encodes for a ubiquitously expressed, mitochondrial GTPase protein. OPA1 has diverse functions within the mitochondrial network, facilitating inner membrane fusion and cristae modelling, regulating mitochondrial DNA maintenance and coordinating mitochondrial bioenergetics. There are currently no licensed disease-modifying therapies for DOA and the disease mechanisms driving RGC degeneration are poorly understood. Here, we describe the generation of isogenic, heterozygous OPA1 null induced pluripotent stem cell (iPSC) (OPA1+/-) through clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 gene editing of a control cell line, in conjunction with the generation of DOA patient-derived iPSC carrying OPA1 variants, namely, the c.2708_2711delTTAG variant (DOA iPSC), and previously reported missense variant iPSC line (c.1334G>A, DOA plus [DOA]+ iPSC) and CRISPR/Cas9 corrected controls. A two-dimensional (2D) differentiation protocol was used to study the effect of OPA1 variants on iPSC-RGC differentiation and mitochondrial function. OPA1+/-, DOA and DOA+ iPSC showed no differentiation deficit compared to control iPSC lines, exhibiting comparable expression of all relevant markers at each stage of differentiation. OPA1+/- and OPA1 variant iPSC-RGCs exhibited impaired mitochondrial homeostasis, with reduced bioenergetic output and compromised mitochondrial DNA maintenance. These data highlight mitochondrial deficits associated with OPA1 dysfunction in human iPSC-RGCs, and establish a platform to study disease mechanisms that contribute to RGC loss in DOA, as well as potential therapeutic interventions.
常染色体显性视神经萎缩(DOA)是最常见的遗传性视神经病变,其特征是视网膜神经节细胞(RGC)的优先丧失,导致视神经变性和进行性双侧中央视力丧失。超过 60%的经基因证实的 DOA 患者携带核 OPA1 基因的变异,该基因编码一种广泛表达的线粒体 GTPase 蛋白。OPA1 在线粒体网络中有多种功能,促进内膜融合和嵴建模,调节线粒体 DNA 维持并协调线粒体生物能量。目前尚无针对 DOA 的许可疾病修饰疗法,并且导致 RGC 变性的疾病机制尚未得到很好的理解。在这里,我们描述了通过使用 CRISPR/Cas9 基因编辑对对照细胞系进行聚类规则间隔短回文重复(CRISPR)/Cas9 基因编辑,生成同基因杂合 OPA1 缺失诱导多能干细胞(iPSC)(OPA1+/-),并结合生成携带 OPA1 变体的 DOA 患者来源的 iPSC,即 c.2708_2711delTTAG 变体(DOA iPSC),以及先前报道的错义变体 iPSC 系(c.1334G>A,DOA plus [DOA]+ iPSC)和 CRISPR/Cas9 校正对照。使用二维(2D)分化方案研究了 OPA1 变体对 iPSC-RGC 分化和线粒体功能的影响。与对照 iPSC 系相比,OPA1+/-、DOA 和 DOA+ iPSC 没有分化缺陷,在分化的每个阶段均表现出所有相关标志物的可比表达。OPA1+/-和 OPA1 变体 iPSC-RGC 表现出受损的线粒体动态平衡,生物能量输出降低,线粒体 DNA 维持受损。这些数据突出了与 OPA1 功能障碍相关的线粒体缺陷在人类 iPSC-RGC 中,为研究导致 DOA 中 RGC 丧失的疾病机制以及潜在的治疗干预措施建立了一个平台。