Department of Botany and Plant Sciences, University of California Reverside, Reverside, California, United States of America.
PLoS One. 2012;7(7):e41336. doi: 10.1371/journal.pone.0041336. Epub 2012 Jul 18.
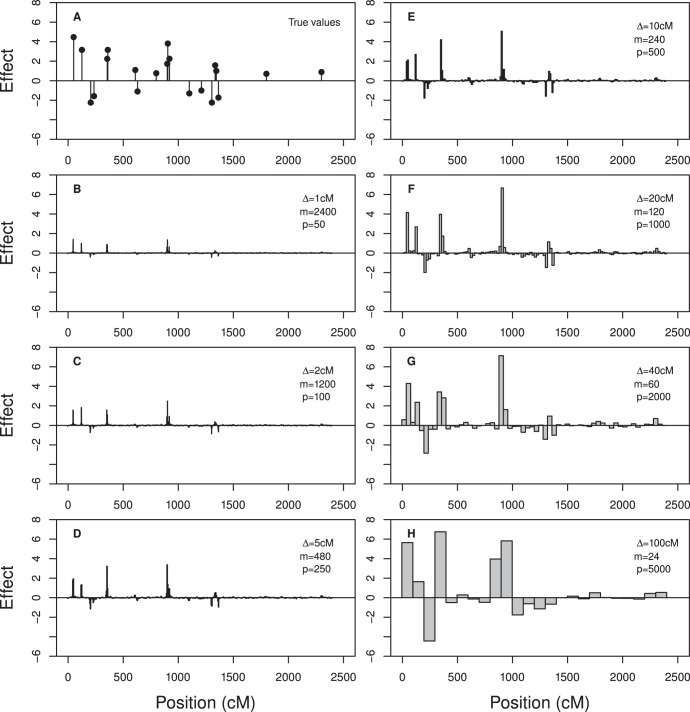
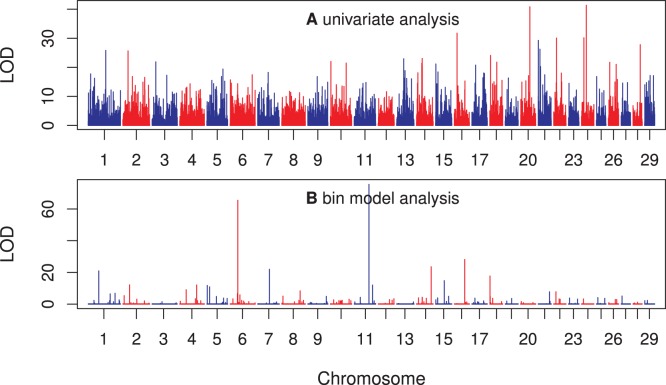
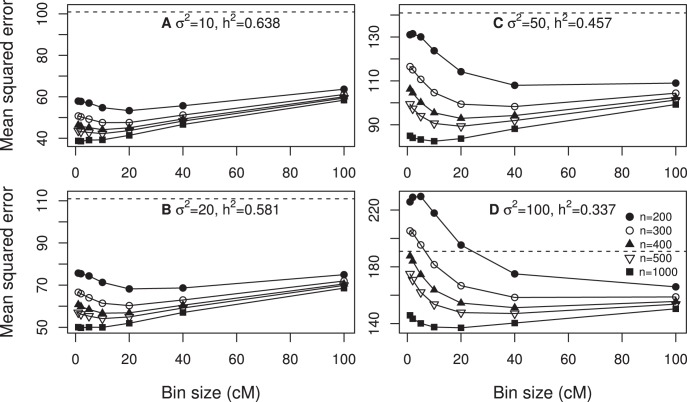
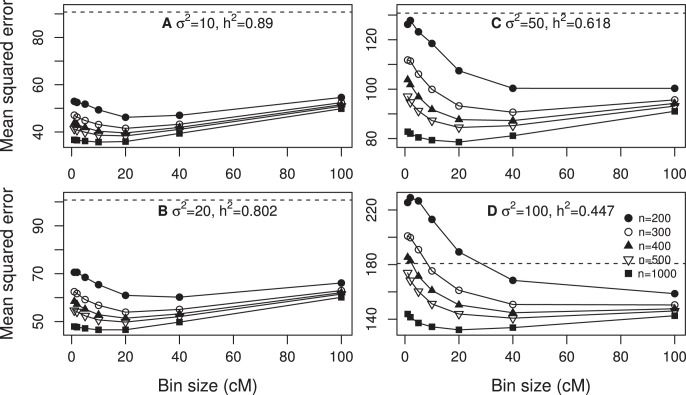
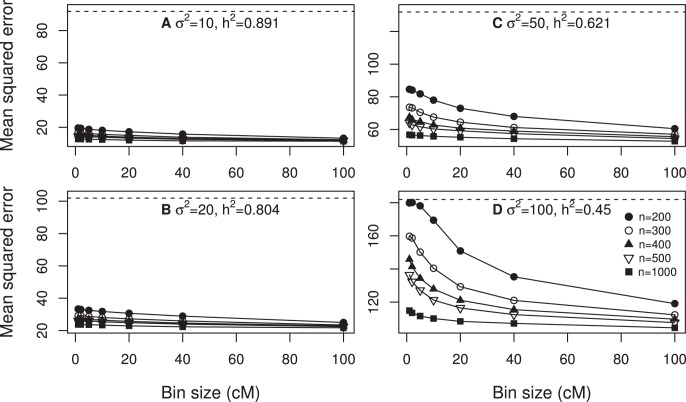
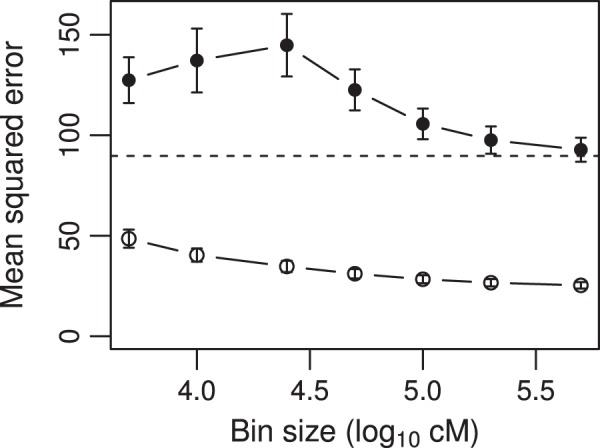
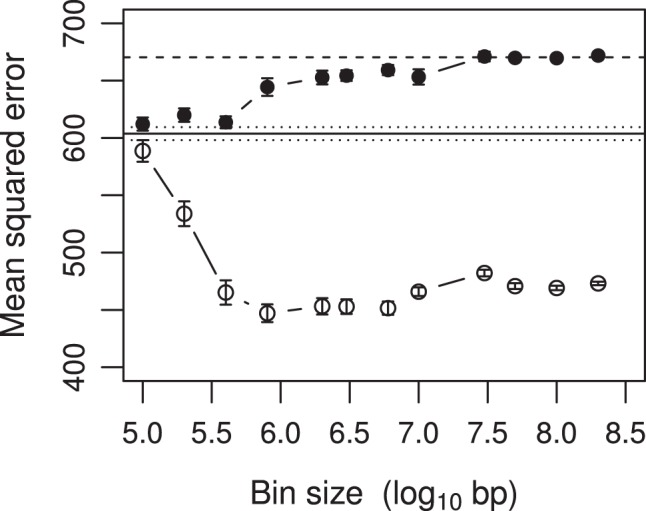
We developed a marker based infinitesimal model for quantitative trait analysis. In contrast to the classical infinitesimal model, we now have new information about the segregation of every individual locus of the entire genome. Under this new model, we propose that the genetic effect of an individual locus is a function of the genome location (a continuous quantity). The overall genetic value of an individual is the weighted integral of the genetic effect function along the genome. Numerical integration is performed to find the integral, which requires partitioning the entire genome into a finite number of bins. Each bin may contain many markers. The integral is approximated by the weighted sum of all the bin effects. We now turn the problem of marker analysis into bin analysis so that the model dimension has decreased from a virtual infinity to a finite number of bins. This new approach can efficiently handle virtually unlimited number of markers without marker selection. The marker based infinitesimal model requires high linkage disequilibrium of all markers within a bin. For populations with low or no linkage disequilibrium, we develop an adaptive infinitesimal model. Both the original and the adaptive models are tested using simulated data as well as beef cattle data. The simulated data analysis shows that there is always an optimal number of bins at which the predictability of the bin model is much greater than the original marker analysis. Result of the beef cattle data analysis indicates that the bin model can increase the predictability from 10% (multiple marker analysis) to 33% (multiple bin analysis). The marker based infinitesimal model paves a way towards the solution of genetic mapping and genomic selection using the whole genome sequence data.
我们开发了一种基于标记的微小模型,用于定量性状分析。与经典的微小模型相比,我们现在有了关于整个基因组中每个个体基因座分离的新信息。在这个新模型下,我们提出个体基因座的遗传效应是基因组位置(连续量)的函数。个体的整体遗传值是沿着基因组的遗传效应函数的加权积分。数值积分用于找到积分,这需要将整个基因组划分为有限数量的箱。每个箱可以包含许多标记。积分通过所有箱效应的加权和来近似。我们现在将标记分析问题转化为箱分析,从而使模型维度从虚拟无限减少到有限数量的箱。这种新方法可以在没有标记选择的情况下高效处理几乎无限数量的标记。基于标记的微小模型要求箱内所有标记的高度连锁不平衡。对于低或无连锁不平衡的群体,我们开发了一种自适应微小模型。原始模型和自适应模型都使用模拟数据和肉牛数据进行了测试。模拟数据分析表明,在最佳箱数下,箱模型的可预测性总是远远大于原始标记分析。肉牛数据分析结果表明,箱模型可以将可预测性从 10%(多标记分析)提高到 33%(多箱分析)。基于标记的微小模型为使用全基因组序列数据进行遗传图谱构建和基因组选择铺平了道路。