Department of Ecology and Evolutionary Biology, Yale University, New Haven, Connecticut, USA.
PLoS One. 2012;7(7):e41250. doi: 10.1371/journal.pone.0041250. Epub 2012 Jul 19.
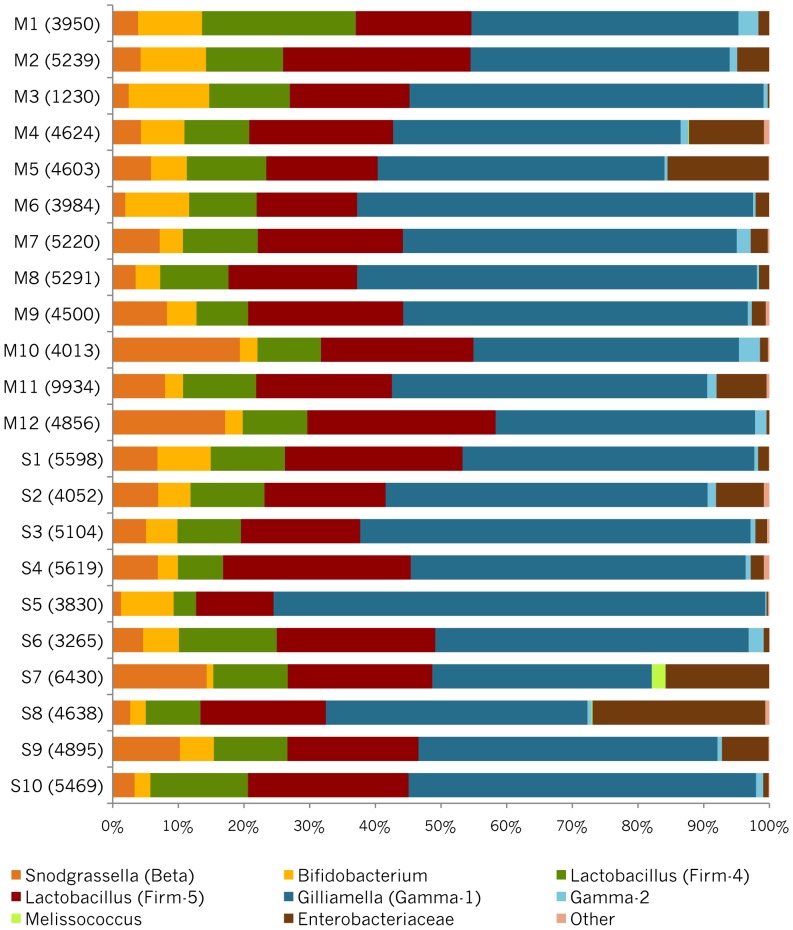
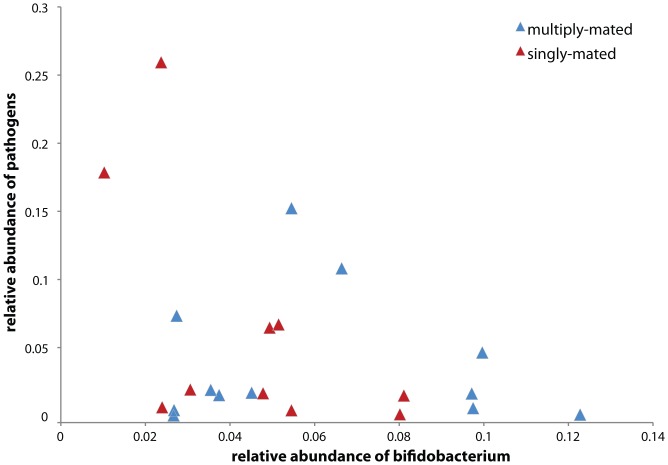
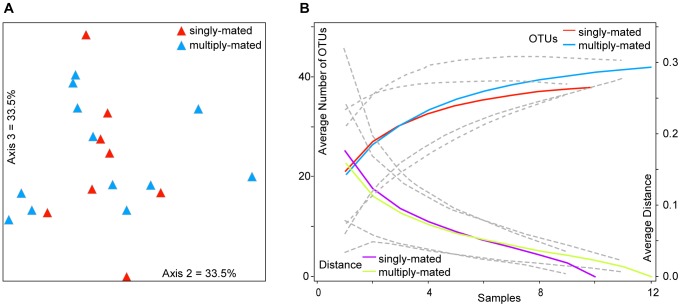
Starting in 2003, numerous studies using culture-independent methodologies to characterize the gut microbiota of honey bees have retrieved a consistent and distinctive set of eight bacterial species, based on near identity of the 16S rRNA gene sequences. A recent study [Mattila HR, Rios D, Walker-Sperling VE, Roeselers G, Newton ILG (2012) Characterization of the active microbiotas associated with honey bees reveals healthier and broader communities when colonies are genetically diverse. PLoS ONE 7(3): e32962], using pyrosequencing of the V1-V2 hypervariable region of the 16S rRNA gene, reported finding entirely novel bacterial species in honey bee guts, and used taxonomic assignments from these reads to predict metabolic activities based on known metabolisms of cultivable species. To better understand this discrepancy, we analyzed the Mattila et al. pyrotag dataset. In contrast to the conclusions of Mattila et al., we found that the large majority of pyrotag sequences belonged to clusters for which representative sequences were identical to sequences from previously identified core species of the bee microbiota. On average, they represent 95% of the bacteria in each worker bee in the Mattila et al. dataset, a slightly lower value than that found in other studies. Some colonies contain small proportions of other bacteria, mostly species of Enterobacteriaceae. Reanalysis of the Mattila et al. dataset also did not support a relationship between abundances of Bifidobacterium and of putative pathogens or a significant difference in gut communities between colonies from queens that were singly or multiply mated. Additionally, consistent with previous studies, the dataset supports the occurrence of considerable strain variation within core species, even within single colonies. The roles of these bacteria within bees, or the implications of the strain variation, are not yet clear.
自 2003 年以来,使用非培养方法研究蜜蜂肠道微生物群的大量研究基于 16S rRNA 基因序列的近同源性,均检索到一致且独特的八细菌种。最近的一项研究[Mattila HR、Rios D、Walker-Sperling VE、Roeselers G、Newton ILG(2012 年)]使用 16S rRNA 基因 V1-V2 高变区的焦磷酸测序,报告发现了蜜蜂肠道中完全新颖的细菌物种,并使用这些读取的分类分配来预测基于可培养物种的已知代谢的代谢活性。为了更好地理解这种差异,我们分析了 Mattila 等人的 pyrotag 数据集。与 Mattila 等人的结论相反,我们发现绝大多数 pyrotag 序列属于与先前鉴定的蜜蜂微生物群核心物种的代表序列完全相同的聚类。平均而言,它们代表了 Mattila 等人数据集每个工蜂中 95%的细菌,略低于其他研究中发现的值。一些蜂群含有少量其他细菌,主要是肠杆菌科的物种。对 Mattila 等人数据集的重新分析也不支持双歧杆菌丰度与假定病原体之间存在关系,或者单配或多配蜂王的蜂群之间肠道群落存在显著差异。此外,与先前的研究一致,该数据集支持核心物种内发生相当大的菌株变异,即使在单个蜂群内也是如此。这些细菌在蜜蜂中的作用或菌株变异的影响尚不清楚。