University of Maryland School of Dentistry, Department of Endodontics, Prosthodontics and Operative Dentistry, Baltimore, MD 20201, USA.
BMC Genomics. 2012 Jul 28;13:345. doi: 10.1186/1471-2164-13-345.
Endodontic infections are a leading cause of oro-facial pain and tooth loss in western countries, and may lead to severe life-threatening infections. These infections are polymicrobial with high bacterial diversity. Understanding the spatial transition of microbiota from normal oral cavities through the infected root canal to the acute periapical abscess can improve our knowledge of the pathogenesis of endodontic infections and lead to more effective treatment. We obtained samples from the oral cavity, infected root canal and periapical abscess of 8 patients (5 with localized and 3 with systemic infections). Microbial populations in these samples were analyzed using next-generation sequencing of 16S rRNA amplicons. Bioinformatics tools and statistical tests with rigorous criteria were used to elucidate the spatial transition of the microbiota from normal to diseased sites.
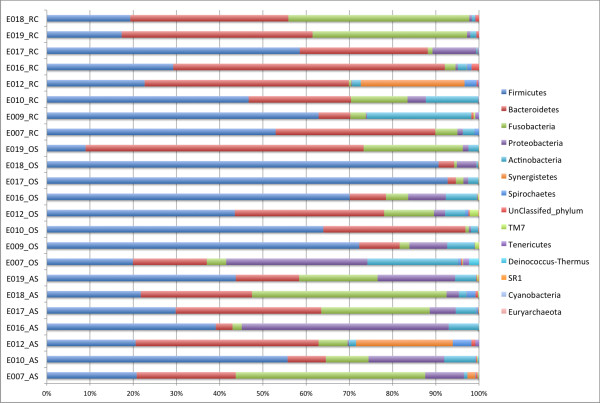
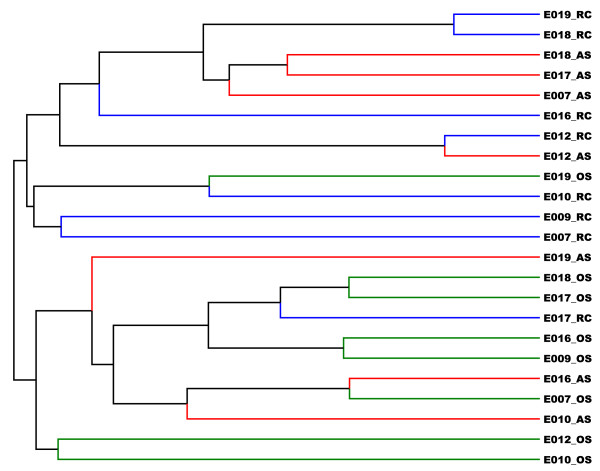
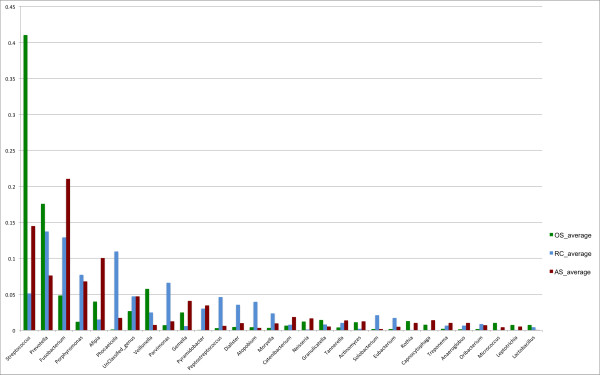
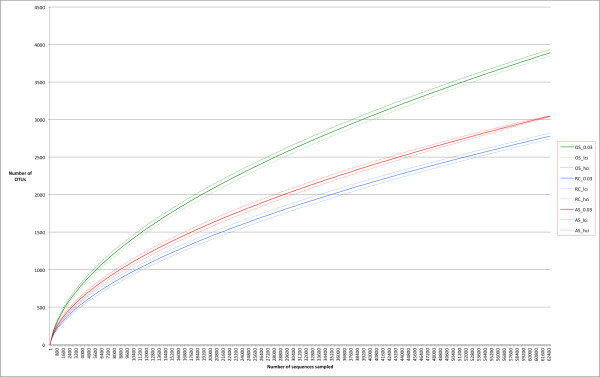
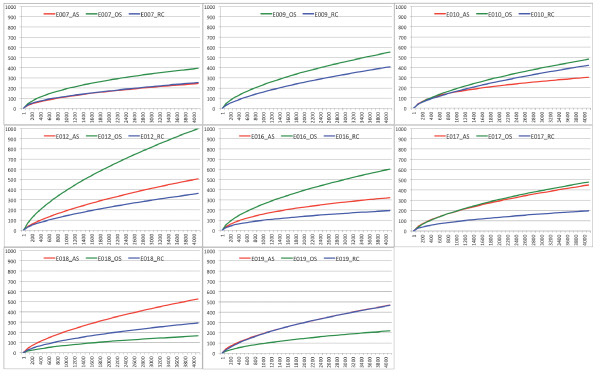
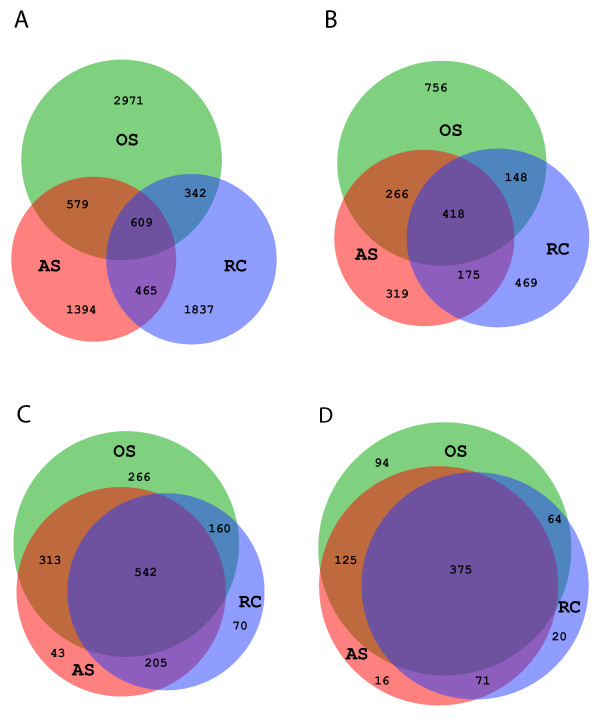
On average, 10,000 partial 16S rRNA gene sequences were obtained from each sample. All sequences fell into 11 different bacterial phyla. The microbial diversity in root canal and abscess samples was significantly lower than in the oral samples. Streptococcus was the most abundant genus in oral cavities while Prevotella and Fusobacterium were most abundant in diseased samples. The microbiota community structures of root canal and abscess samples were, however, more similar to each other than to the oral cavity microbiota. Using rigorous criteria and novel bioinformatics tools, we found that Granulicatella adiacens, Eubacterium yurii, Prevotella melaninogenica, Prevotella salivae, Streptococcus mitis, and Atopobium rimae were over-represented in diseased samples.
We used a novel approach and high-throughput methodologies to characterize the microbiota associated normal and diseased oral sites in the same individuals.
牙髓感染是西方国家口腔颌面部疼痛和牙齿缺失的主要原因,可能导致严重的危及生命的感染。这些感染是多微生物的,具有高度的细菌多样性。了解微生物群从正常口腔通过感染的根管到急性根尖脓肿的空间转移,可以提高我们对牙髓感染发病机制的认识,并导致更有效的治疗。我们从 8 名患者(5 名局部感染和 3 名全身感染)的口腔、感染根管和根尖脓肿中获得样本。使用 16S rRNA 扩增子的下一代测序分析这些样本中的微生物种群。使用严格标准的生物信息学工具和统计检验来阐明微生物群从正常到患病部位的空间转移。
平均而言,从每个样本中获得了 10,000 个 16S rRNA 基因的部分序列。所有序列分为 11 个不同的细菌门。根管和脓肿样本中的微生物多样性明显低于口腔样本。链球菌是口腔中最丰富的属,而普雷沃氏菌和梭杆菌在患病样本中最丰富。然而,根管和脓肿样本的微生物群落结构彼此之间比与口腔微生物群落更相似。使用严格的标准和新颖的生物信息学工具,我们发现颗粒性放线杆菌、尤氏杆菌、黑色素普雷沃氏菌、唾液普雷沃氏菌、链球菌和口咽拟杆菌在患病样本中过度表达。
我们使用了一种新的方法和高通量方法来描述同一个体正常和患病口腔部位相关的微生物群。