Department of Biological Sciences, University of Helsinki, PL 65, Viikinkaari 1, 00014, Helsinki, Finland.
BMC Genomics. 2012 Aug 1;13:361. doi: 10.1186/1471-2164-13-361.
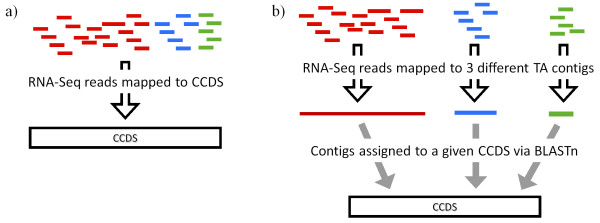
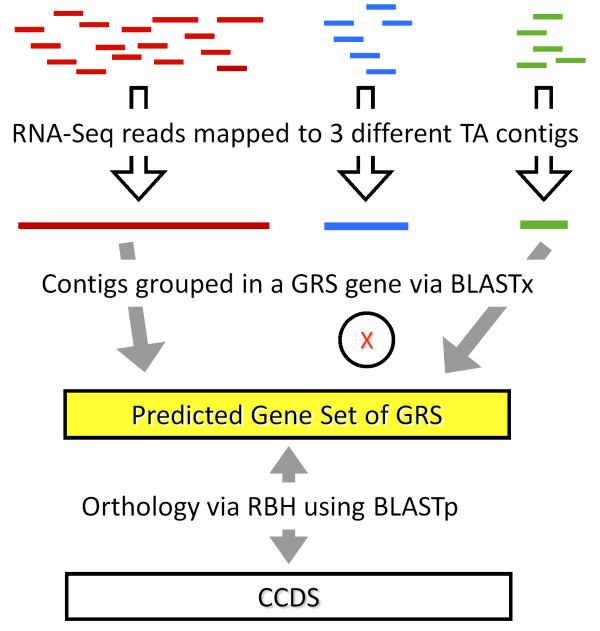
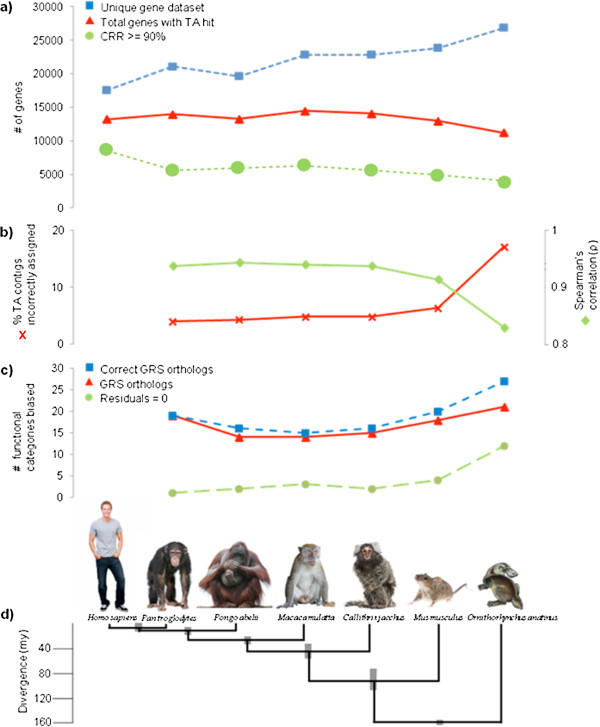
How well does RNA-Seq data perform for quantitative whole gene expression analysis in the absence of a genome? This is one unanswered question facing the rapidly growing number of researchers studying non-model species. Using Homo sapiens data and resources, we compared the direct mapping of sequencing reads to predicted genes from the genome with mapping to de novo transcriptomes assembled from RNA-Seq data. Gene coverage and expression analysis was further investigated in the non-model context by using increasingly divergent genomic reference species to group assembled contigs by unique genes.
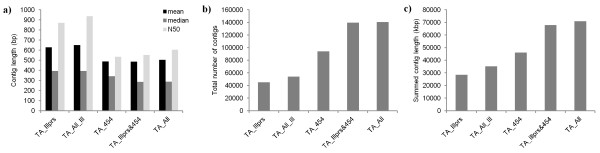
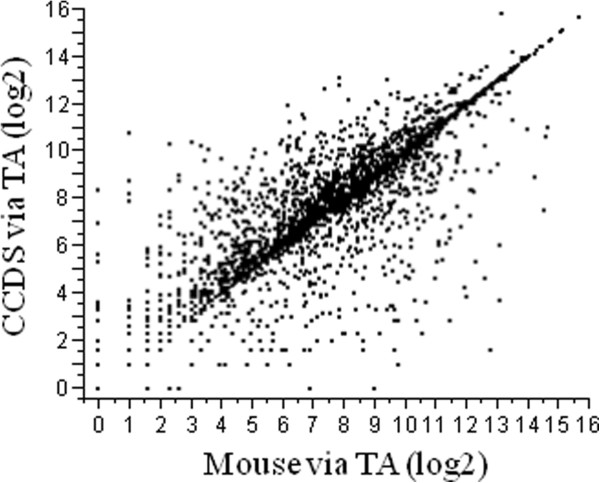
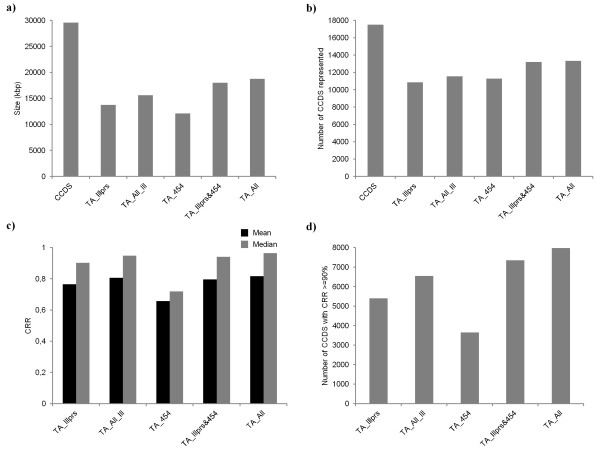
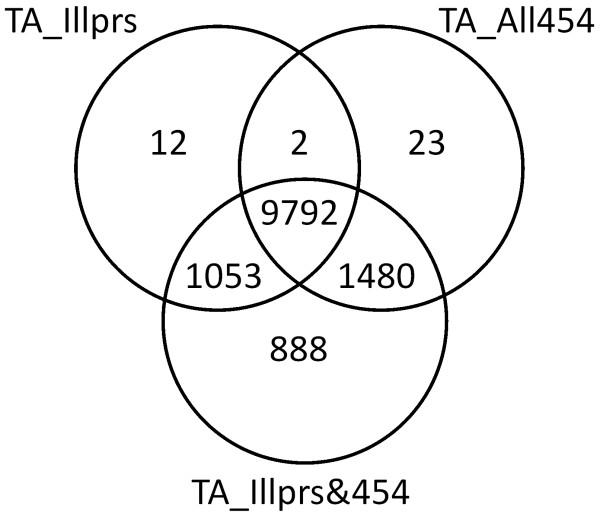
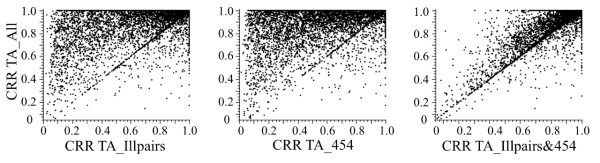
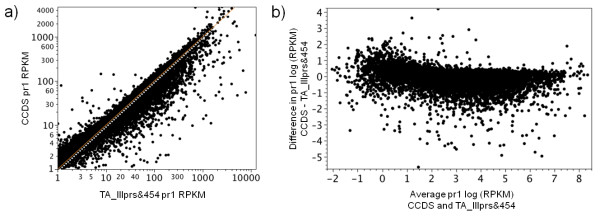
Eight transcriptome sets, composed of varying amounts of Illumina and 454 data, were assembled and assessed. Hybrid 454/Illumina assemblies had the highest transcriptome and individual gene coverage. Quantitative whole gene expression levels were highly similar between using a de novo hybrid assembly and the predicted genes as a scaffold, although mapping to the de novo transcriptome assembly provided data on fewer genes. Using non-target species as reference scaffolds does result in some loss of sequence and expression data, and bias and error increase with evolutionary distance. However, within a 100 million year window these effect sizes are relatively small.
Predicted gene sets from sequenced genomes of related species can provide a powerful method for grouping RNA-Seq reads and annotating contigs. Gene expression results can be produced that are similar to results obtained using gene models derived from a high quality genome, though biased towards conserved genes. Our results demonstrate the power and limitations of conducting RNA-Seq in non-model species.
在缺乏基因组的情况下,RNA-Seq 数据在定量全基因表达分析方面的表现如何?这是一个尚未解决的问题,目前有越来越多的研究非模式物种的研究人员面临这个问题。利用智人数据和资源,我们比较了测序reads 直接映射到基因组中预测基因与从 RNA-Seq 数据组装的从头转录组之间的映射。通过使用越来越多的基因组参考物种,将组装的 contigs 按独特基因分组,进一步在非模型背景下研究了基因覆盖和表达分析。
组装并评估了 8 个转录组集,其中包含不同数量的 Illumina 和 454 数据。杂交 454/Illumina 组装具有最高的转录组和单个基因覆盖度。使用从头杂交组装和预测基因作为支架进行定量全基因表达水平非常相似,尽管映射到从头转录组组装提供了更少的基因数据。使用非目标物种作为参考支架确实会导致一些序列和表达数据的丢失,并且随着进化距离的增加,偏差和错误会增加。然而,在 1 亿年的窗口内,这些效应大小相对较小。
来自相关物种测序基因组的预测基因集可以为分组 RNA-Seq 读取和注释 contigs 提供一种强大的方法。可以产生类似于使用高质量基因组衍生的基因模型获得的基因表达结果,但偏向于保守基因。我们的结果展示了在非模式物种中进行 RNA-Seq 的优势和局限性。