Laboratory of Malariology, Research Institute for Microbial Diseases, Osaka University, Suita, Japan.
Nat Genet. 2012 Sep;44(9):1051-5. doi: 10.1038/ng.2375. Epub 2012 Aug 5.
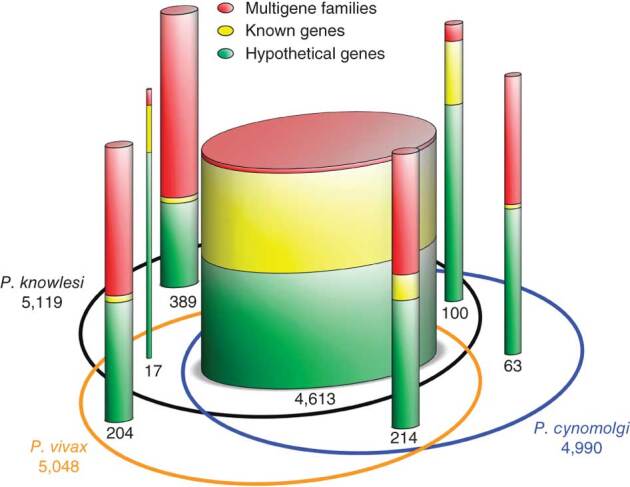
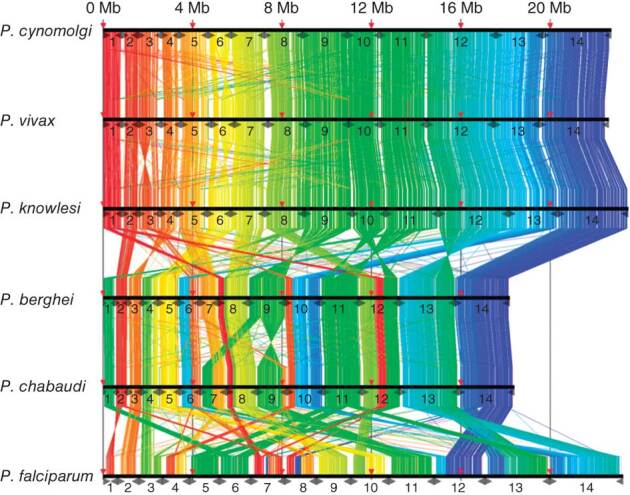
P. cynomolgi, a malaria-causing parasite of Asian Old World monkeys, is the sister taxon of P. vivax, the most prevalent malaria-causing species in humans outside of Africa. Because P. cynomolgi shares many phenotypic, biological and genetic characteristics with P. vivax, we generated draft genome sequences for three P. cynomolgi strains and performed genomic analysis comparing them with the P. vivax genome, as well as with the genome of a third previously sequenced simian parasite, Plasmodium knowlesi. Here, we show that genomes of the monkey malaria clade can be characterized by copy-number variants (CNVs) in multigene families involved in evasion of the human immune system and invasion of host erythrocytes. We identify genome-wide SNPs, microsatellites and CNVs in the P. cynomolgi genome, providing a map of genetic variation that can be used to map parasite traits and study parasite populations. The sequencing of the P. cynomolgi genome is a critical step in developing a model system for P. vivax research and in counteracting the neglect of P. vivax.
食蟹猴疟原虫是一种导致亚洲旧世界猴发生疟疾的寄生虫,是人类中非洲以外最常见疟原虫——间日疟原虫的姊妹分类群。由于食蟹猴疟原虫与间日疟原虫在表型、生物学和遗传学特征上有许多相似之处,因此我们生成了 3 株食蟹猴疟原虫株的草图基因组序列,并进行了基因组分析,将其与间日疟原虫基因组以及之前测序的第三种灵长类寄生虫疟原虫 knowlesi 的基因组进行了比较。在这里,我们表明,猴疟原虫类群的基因组可以通过参与逃避人体免疫系统和宿主红细胞入侵的多基因家族中的拷贝数变异(CNVs)来进行特征描述。我们在食蟹猴疟原虫基因组中鉴定出了全基因组 SNP、微卫星和 CNVs,为遗传变异图谱提供了一个图谱,可用于定位寄生虫特征和研究寄生虫种群。食蟹猴疟原虫基因组的测序是开发间日疟原虫研究模型系统和对抗间日疟原虫被忽视的关键步骤。