Key Laboratory of Crop Gene Resources and Germplasm Enhancement, Ministry of Agriculture, Institute of Crop Science, Chinese Academy of Agricultural Sciences, Zhongguancun, Beijing, People's Republic of China.
BMC Genomics. 2012 Aug 14;13:392. doi: 10.1186/1471-2164-13-392.
Rapid advances in next-generation sequencing methods have provided new opportunities for transcriptome sequencing (RNA-Seq). The unprecedented sequencing depth provided by RNA-Seq makes it a powerful and cost-efficient method for transcriptome study, and it has been widely used in model organisms and non-model organisms to identify and quantify RNA. For non-model organisms lacking well-defined genomes, de novo assembly is typically required for downstream RNA-Seq analyses, including SNP discovery and identification of genes differentially expressed by phenotypes. Although RNA-Seq has been successfully used to sequence many non-model organisms, the results of de novo assembly from short reads can still be improved by using recent bioinformatic developments.
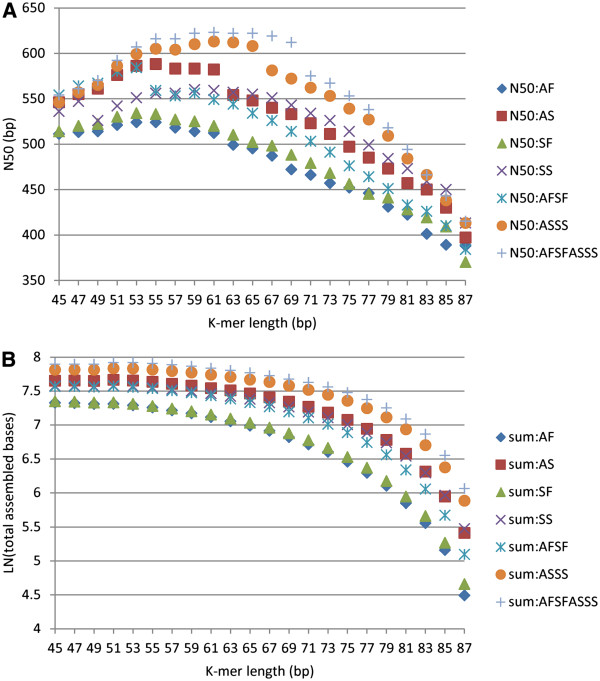
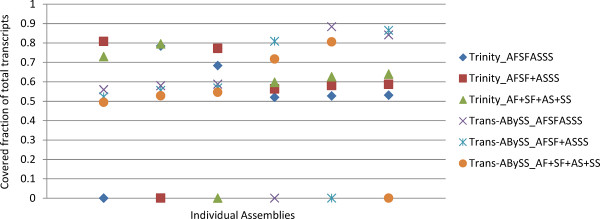
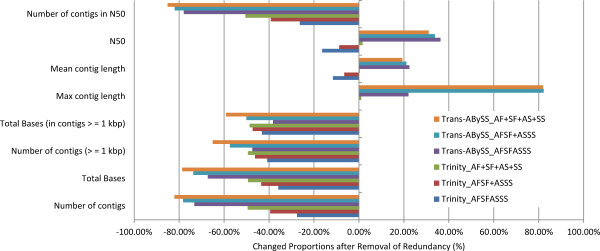
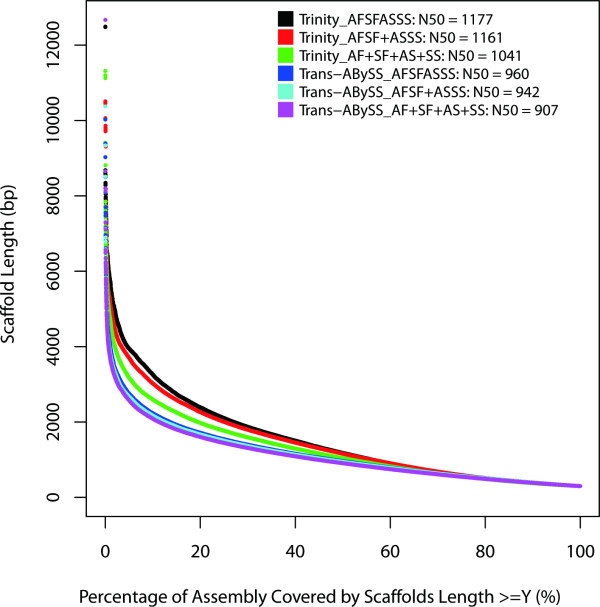
In this study, we used 212.6 million pair-end reads, which accounted for 16.2 Gb, to assemble the hexaploid wheat transcriptome. Two state-of-the-art assemblers, Trinity and Trans-ABySS, which use the single and multiple k-mer methods, respectively, were used, and the whole de novo assembly process was divided into the following four steps: pre-assembly, merging different samples, removal of redundancy and scaffolding. We documented every detail of these steps and how these steps influenced assembly performance to gain insight into transcriptome assembly from short reads. After optimization, the assembled transcripts were comparable to Sanger-derived ESTs in terms of both continuity and accuracy. We also provided considerable new wheat transcript data to the community.
It is feasible to assemble the hexaploid wheat transcriptome from short reads. Special attention should be paid to dealing with multiple samples to balance the spectrum of expression levels and redundancy. To obtain an accurate overview of RNA profiling, removal of redundancy may be crucial in de novo assembly.
下一代测序方法的快速发展为转录组测序(RNA-Seq)提供了新的机会。RNA-Seq 提供的前所未有的测序深度使其成为转录组研究的强大且经济高效的方法,已广泛应用于模式生物和非模式生物,用于鉴定和定量 RNA。对于缺乏明确基因组的非模式生物,通常需要进行从头组装,以便进行下游 RNA-Seq 分析,包括 SNP 发现和表型差异表达基因的鉴定。尽管 RNA-Seq 已成功用于许多非模式生物的测序,但通过使用最新的生物信息学进展,仍可以改善来自短读长的从头组装结果。
本研究使用了 2.126 亿对末端读长,总计 16.2GB,组装了六倍体小麦转录组。使用了两种最先进的组装器,Trinity 和 Trans-ABySS,分别使用单和多 k-mer 方法,整个从头组装过程分为以下四个步骤:预组装、合并不同的样本、去除冗余和支架搭建。我们记录了这些步骤的每一个细节以及这些步骤如何影响组装性能,以深入了解来自短读长的转录组组装。经过优化,组装的转录本在连续性和准确性方面与 Sanger 衍生的 EST 相当。我们还为社区提供了大量新的小麦转录数据。
从短读长组装六倍体小麦转录组是可行的。应特别注意处理多个样本,以平衡表达水平和冗余的分布。为了获得 RNA 谱的准确概述,在从头组装中去除冗余可能至关重要。