Department of Microbiology, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, Pennsylvania, United States of America.
PLoS One. 2012;7(8):e42342. doi: 10.1371/journal.pone.0042342. Epub 2012 Aug 10.
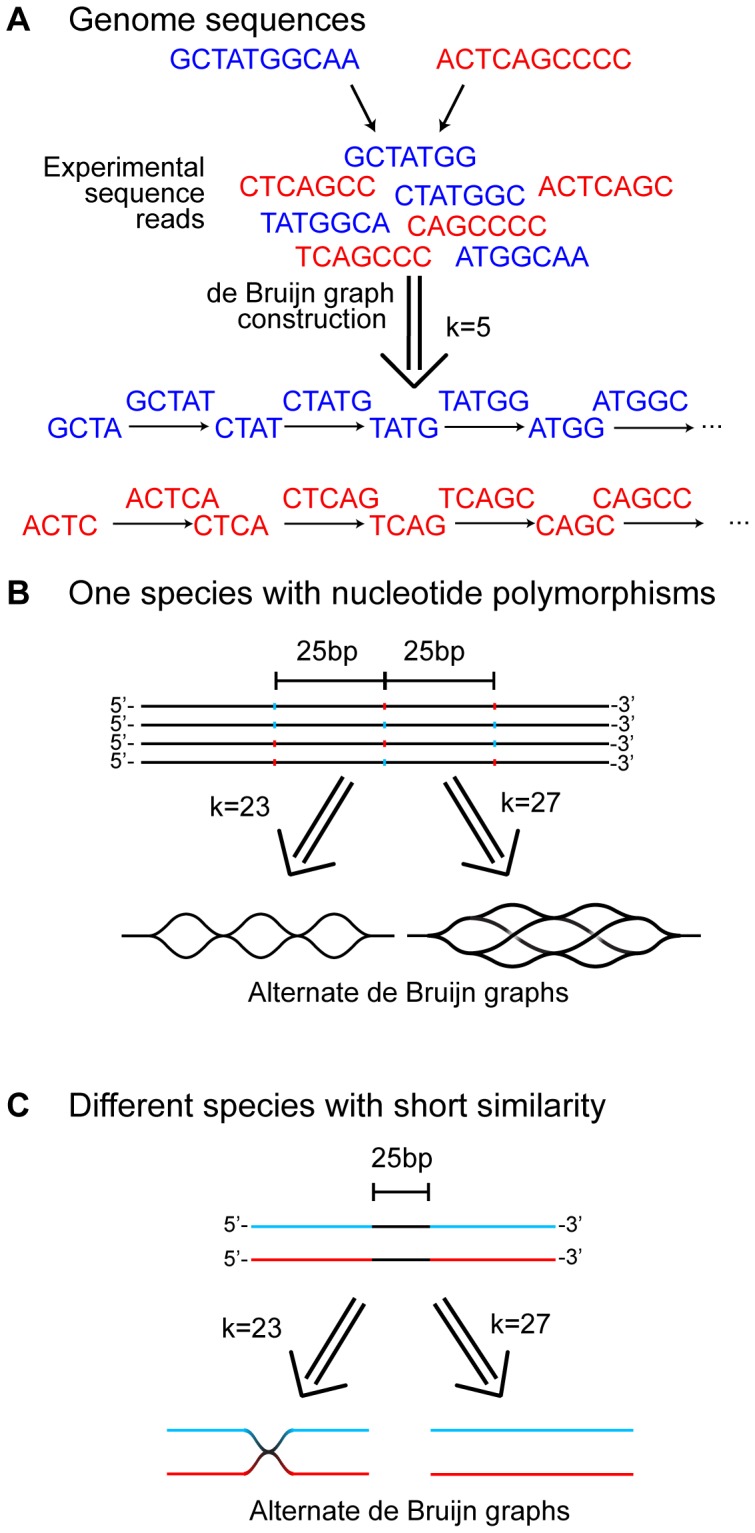
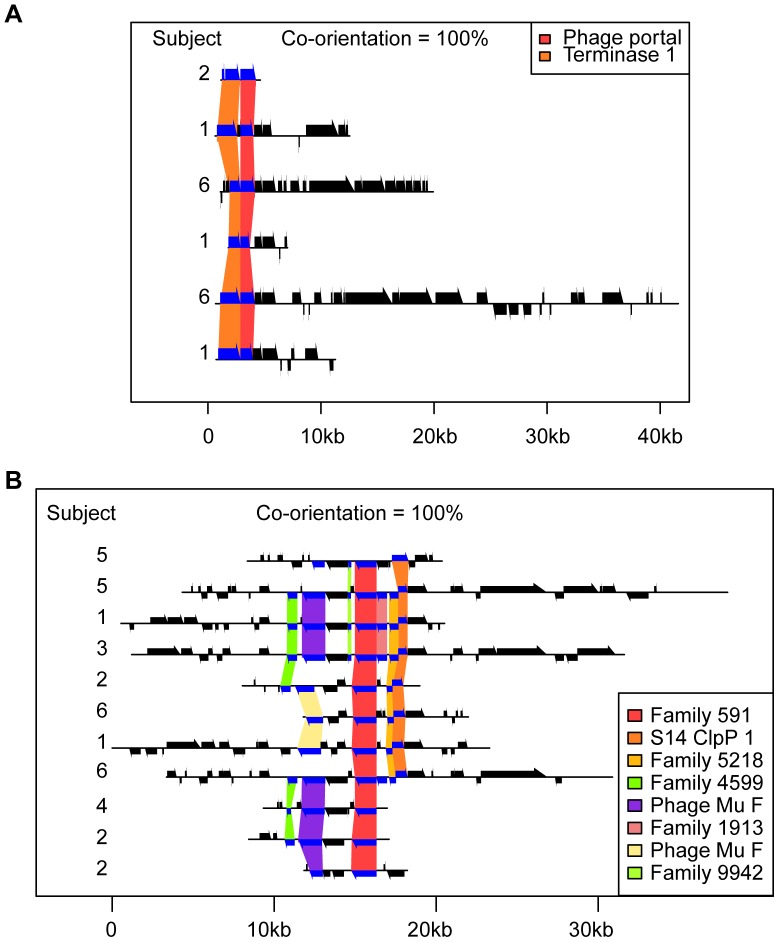
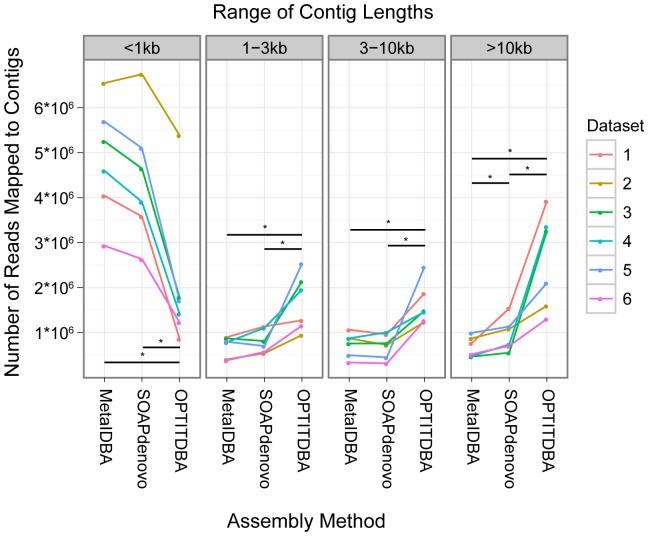
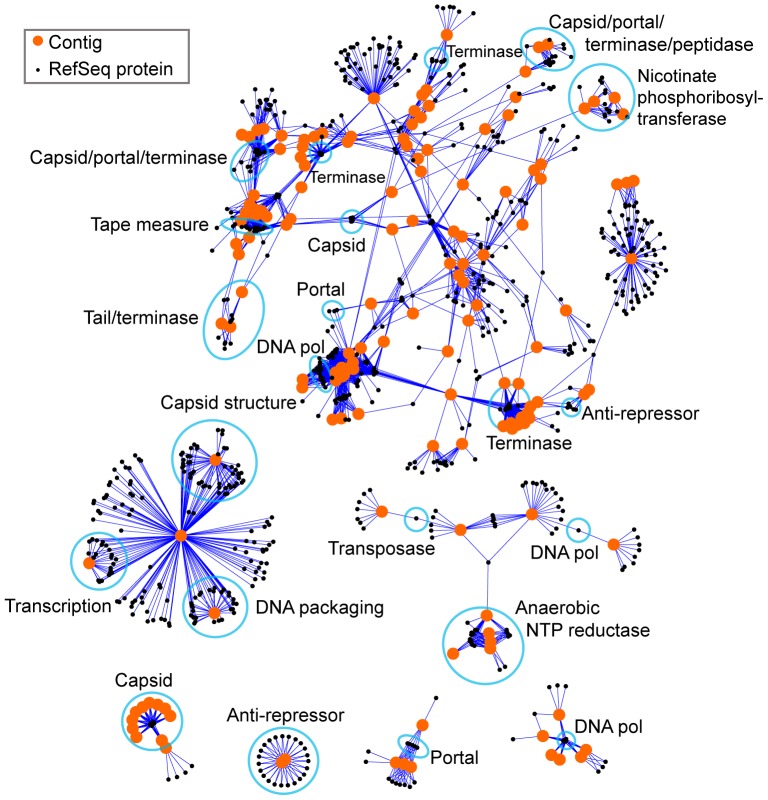
Viruses are a crucial component of the human microbiome, but large population sizes, high sequence diversity, and high frequencies of novel genes have hindered genomic analysis by high-throughput sequencing. Here we investigate approaches to metagenomic assembly to probe genome structure in a sample of 5.6 Gb of gut viral DNA sequence from six individuals. Tests showed that a new pipeline based on DeBruijn graph assembly yielded longer contigs that were able to recruit more reads than the equivalent non-optimized, single-pass approach. To characterize gene content, the database of viral RefSeq proteins was compared to the assembled viral contigs, generating a bipartite graph with functional cassettes linking together viral contigs, which revealed a high degree of connectivity between diverse genomes involving multiple genes of the same functional class. In a second step, open reading frames were grouped by their co-occurrence on contigs in a database-independent manner, revealing conserved cassettes of co-oriented ORFs. These methods reveal that free-living bacteriophages, while usually dissimilar at the nucleotide level, often have significant similarity at the level of encoded amino acid motifs, gene order, and gene orientation. These findings thus connect contemporary metagenomic analysis with classical studies of bacteriophage genomic cassettes. Software is available at https://sourceforge.net/projects/optitdba/.
病毒是人类微生物组的重要组成部分,但由于种群规模大、序列多样性高、新基因频率高,高通量测序的基因组分析受到了阻碍。在这里,我们研究了宏基因组组装的方法,以探测来自六个人的 56 亿碱基对肠道病毒 DNA 序列样本中的基因组结构。测试表明,基于 DeBruijn 图组装的新管道产生的 contigs 比等效的非优化单步方法能够招募更多的读取。为了描述基因组成,将病毒 RefSeq 蛋白数据库与组装的病毒 contigs 进行比较,生成了一个具有功能盒的二分图,将病毒 contigs 连接在一起,揭示了不同基因组之间高度的连通性,涉及同一功能类别的多个基因。在第二步中,以数据库独立的方式将开放阅读框按其在 contigs 上的共现进行分组,揭示了共取向 ORFs 的保守盒。这些方法表明,自由生活的噬菌体虽然在核苷酸水平上通常不同,但在编码的氨基酸基序、基因顺序和基因取向方面往往具有显著的相似性。这些发现将当代宏基因组分析与噬菌体基因组盒的经典研究联系起来。软件可在 https://sourceforge.net/projects/optitdba/ 获得。