Graduate Group in Biophysics, University of California, San Francisco, San Francisco, California, United States of America.
PLoS Comput Biol. 2012;8(8):e1002639. doi: 10.1371/journal.pcbi.1002639. Epub 2012 Aug 23.
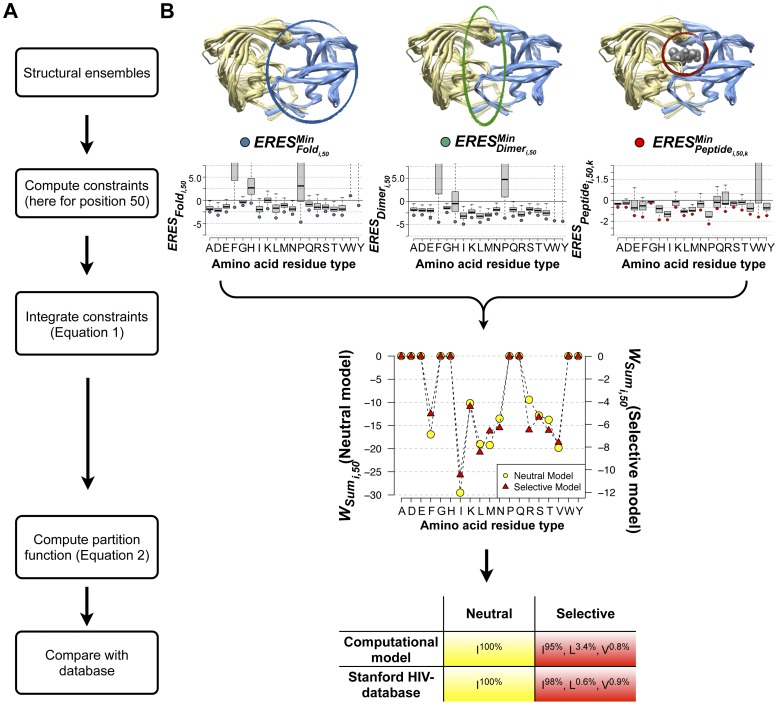
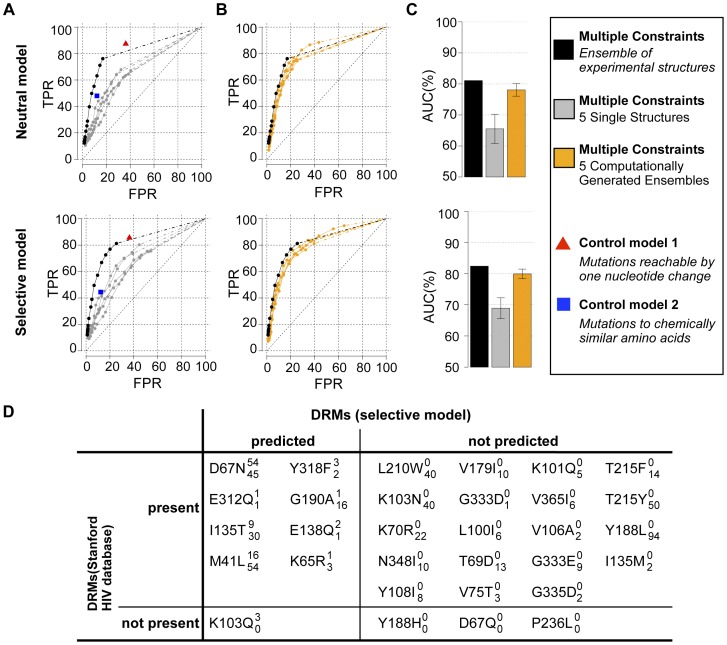
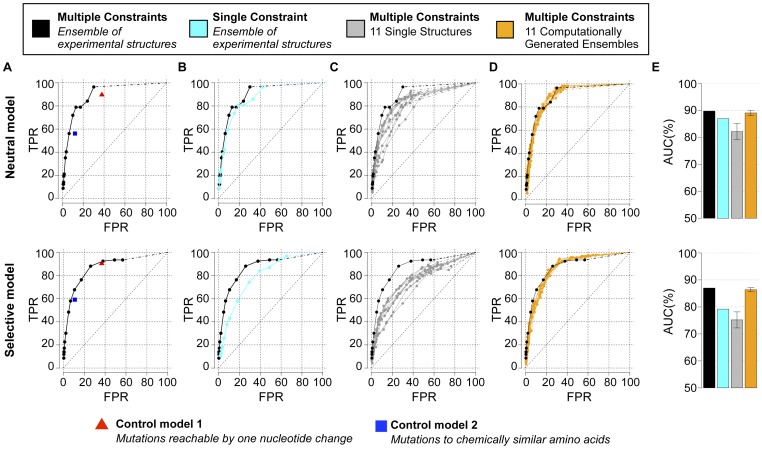
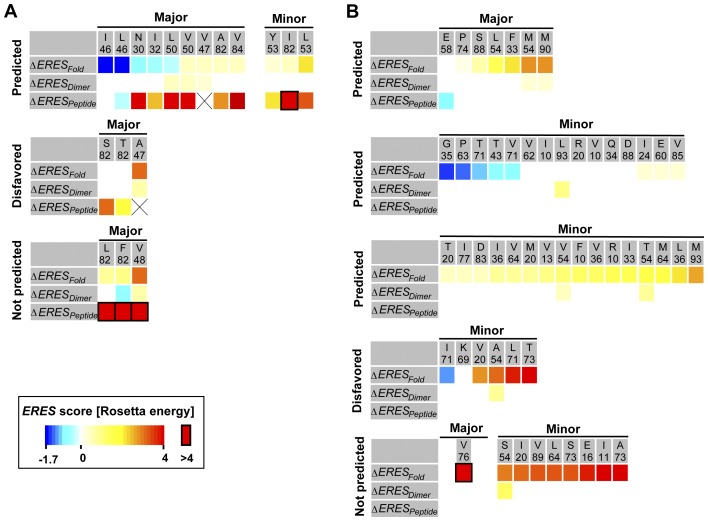
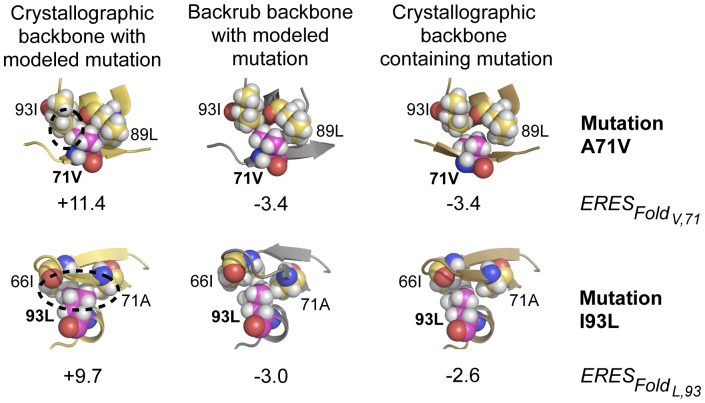
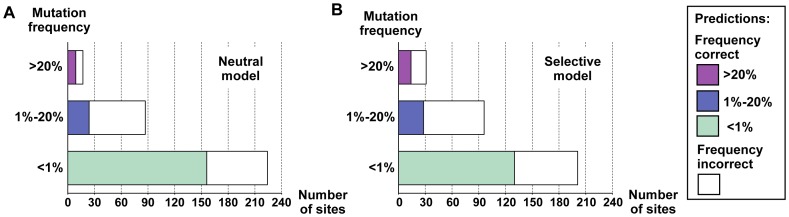
Predicting which mutations proteins tolerate while maintaining their structure and function has important applications for modeling fundamental properties of proteins and their evolution; it also drives progress in protein design. Here we develop a computational model to predict the tolerated sequence space of HIV-1 protease reachable by single mutations. We assess the model by comparison to the observed variability in more than 50,000 HIV-1 protease sequences, one of the most comprehensive datasets on tolerated sequence space. We then extend the model to a second protein, reverse transcriptase. The model integrates multiple structural and functional constraints acting on a protein and uses ensembles of protein conformations. We find the model correctly captures a considerable fraction of protease and reverse-transcriptase mutational tolerance and shows comparable accuracy using either experimentally determined or computationally generated structural ensembles. Predictions of tolerated sequence space afforded by the model provide insights into stability-function tradeoffs in the emergence of resistance mutations and into strengths and limitations of the computational model.
预测蛋白质在保持其结构和功能的同时能容忍哪些突变,对于模拟蛋白质的基本性质及其进化具有重要意义;它也推动了蛋白质设计的进展。在这里,我们开发了一种计算模型来预测 HIV-1 蛋白酶单突变可达到的耐受序列空间。我们通过将模型与超过 50000 个 HIV-1 蛋白酶序列的观察到的可变性进行比较来评估模型,这是关于耐受序列空间的最全面的数据集之一。然后,我们将模型扩展到第二种蛋白质,逆转录酶。该模型整合了作用于蛋白质的多种结构和功能约束,并使用蛋白质构象的集合。我们发现该模型正确地捕获了相当一部分蛋白酶和逆转录酶的突变耐受性,并且使用实验确定或计算生成的结构集合都具有相当的准确性。该模型提供的耐受序列空间预测为耐药突变出现时的稳定性-功能权衡以及计算模型的优缺点提供了深入了解。