Center for Structural Biology, Vanderbilt University, Nashville, Tennessee, United States of America.
Vanderbilt Vaccine Center, Vanderbilt University Medical Center, Nashville, Tennessee, United States of America.
PLoS Comput Biol. 2020 Feb 7;16(2):e1007339. doi: 10.1371/journal.pcbi.1007339. eCollection 2020 Feb.
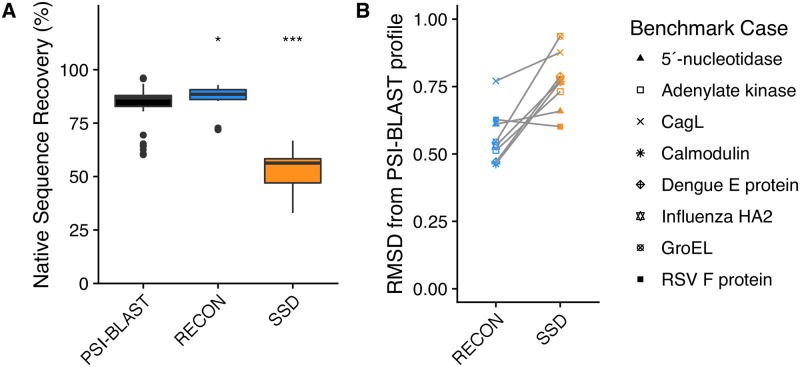
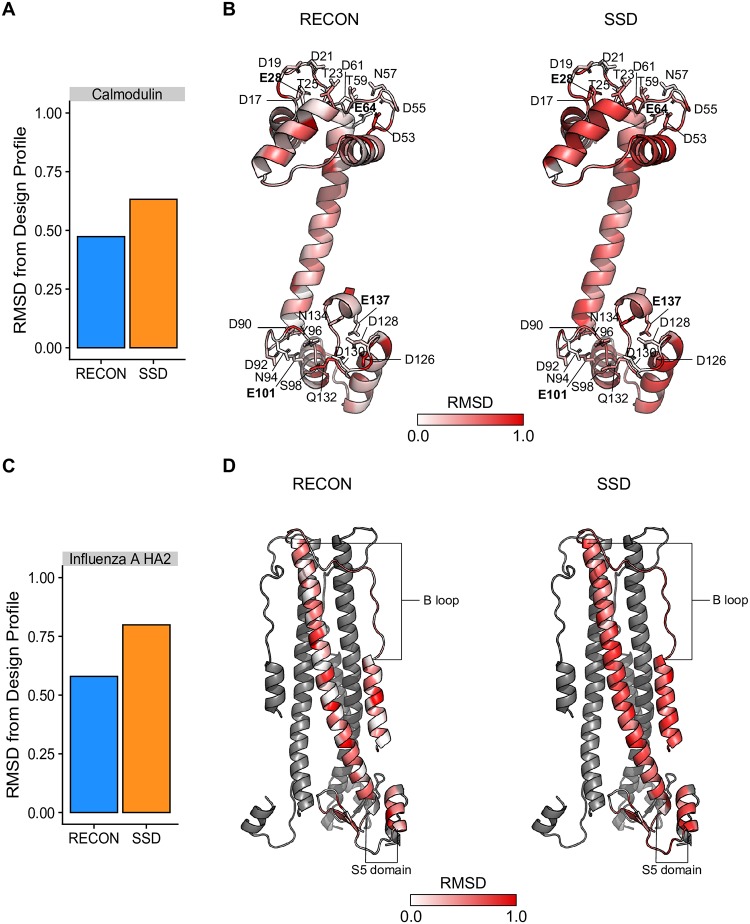
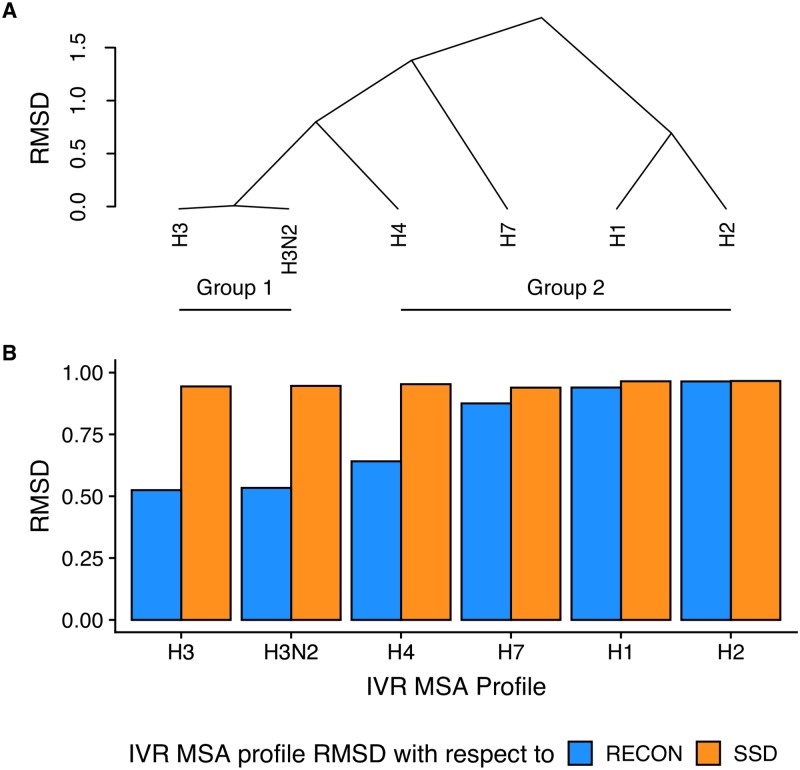
Computational protein design of an ensemble of conformations for one protein-i.e., multi-state design-determines the side chain identity by optimizing the energetic contributions of that side chain in each of the backbone conformations. Sampling the resulting large sequence-structure search space limits the number of conformations and the size of proteins in multi-state design algorithms. Here, we demonstrated that the REstrained CONvergence (RECON) algorithm can simultaneously evaluate the sequence of large proteins that undergo substantial conformational changes. Simultaneous optimization of side chain conformations across all conformations increased sequence conservation when compared to single-state designs in all cases. More importantly, the sequence space sampled by RECON MSD resembled the evolutionary sequence space of flexible proteins, particularly when confined to predicting the mutational preferences of limited common ancestral descent, such as in the case of influenza type A hemagglutinin. Additionally, we found that sequence positions which require substantial changes in their local environment across an ensemble of conformations are more likely to be conserved. These increased conservation rates are better captured by RECON MSD over multiple conformations and thus multiple local residue environments during design. To quantify this rewiring of contacts at a certain position in sequence and structure, we introduced a new metric designated 'contact proximity deviation' that enumerates contact map changes. This measure allows mapping of global conformational changes into local side chain proximity adjustments, a property not captured by traditional global similarity metrics such as RMSD or local similarity metrics such as changes in φ and ψ angles.
对一个蛋白质的构象集合进行计算蛋白质设计,即多态性设计,通过优化该侧链在每个骨架构象中的能量贡献来确定侧链的身份。对由此产生的大序列-结构搜索空间进行采样限制了多态性设计算法中构象的数量和蛋白质的大小。在这里,我们证明了受限收敛(RECON)算法可以同时评估经历大量构象变化的大蛋白质的序列。与单态设计相比,在所有情况下,对所有构象的侧链构象进行同时优化都会增加序列保守性。更重要的是,RECON MSD 采样的序列空间与柔性蛋白质的进化序列空间相似,特别是当限制为预测有限共同祖先的突变偏好时,例如在甲型流感血凝素的情况下。此外,我们发现,在构象集合中,需要在其局部环境中发生大量变化的序列位置更有可能保持保守。RECON MSD 在多个构象和设计过程中的多个局部残基环境中更好地捕捉到这种接触的重新布线。为了量化序列和结构中某个位置的接触重新布线,我们引入了一个新的度量标准,称为“接触接近偏差”,它可以枚举接触图的变化。该度量标准允许将全局构象变化映射到局部侧链接近调整,这是传统全局相似性度量(如 RMSD)或局部相似性度量(如 φ 和 ψ 角的变化)无法捕捉到的属性。