Molecular and Computational Biology, University of Southern California, Los Angeles, CA 90089, USA.
Nucleic Acids Res. 2013 Jan 7;41(1):e7. doi: 10.1093/nar/gks800. Epub 2012 Aug 31.
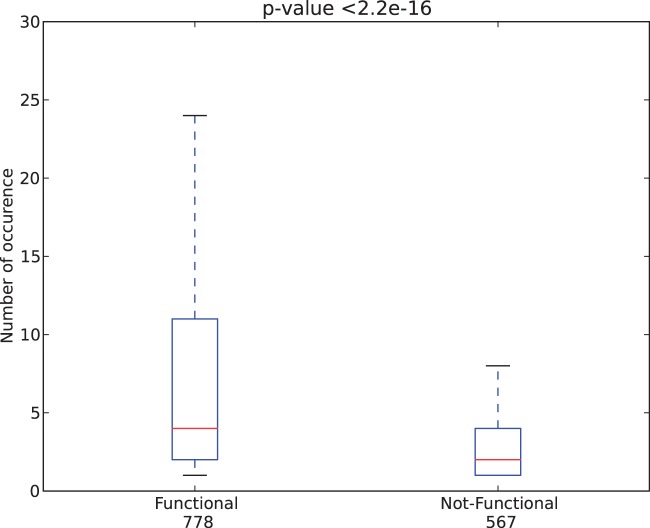
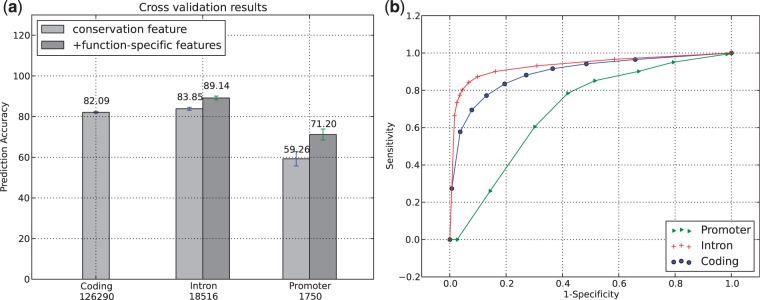
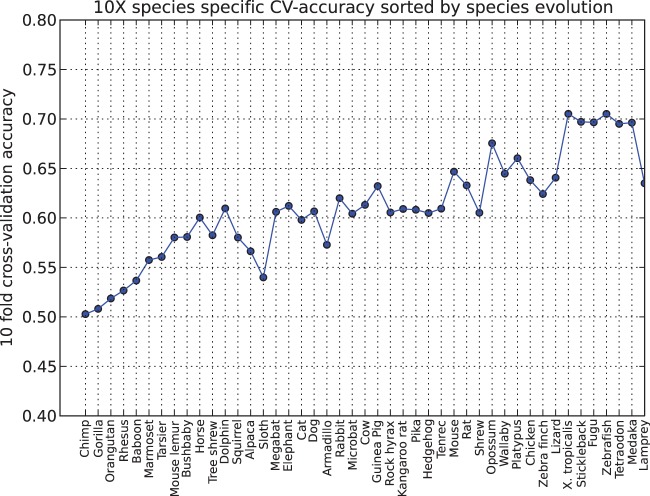
The thousand genomes project and many similar ongoing large-scale sequencing efforts require new methods to predict functional variants in both coding and non-coding regions in order to understand phenotype and genotype relationships. We report the design of a new model SInBaD (Sequence-Information-Based-Decision-model) which relies on nucleotide conservation information to evaluate any annotated human variant in all known exons, introns, splice junctions and promoter regions. SInBaD builds separate mathematical models for promoters, exons and introns, using the human disease mutations annotated in human gene mutation database as the training dataset for functional variants. The ten-fold cross validation shows high prediction accuracy. Validations on test datasets, demonstrate that variants predicted as functional have a significantly higher occurrence in cancer patients. We also applied our model to variants found in four different individual human genomes to identify a set of functional variants, which might be of interest for further studies. Scores for any possible variants for all annotated genes are available under http://tingchenlab.cmb.usc.edu/sinbad/. SInBaD supports the current standard format of genotyping, the variant call files (VCF 4.0), making it easy to integrate it into any existing next-generation sequencing pipeline. The accuracy of SNP detection poses the only limitation to the use of SInBaD.
千基因组计划和许多类似的正在进行的大规模测序工作需要新的方法来预测编码和非编码区域中的功能变体,以便理解表型和基因型的关系。我们报告了一种新模型 SInBaD(基于序列信息的决策模型)的设计,该模型依赖于核苷酸保守性信息来评估所有已知外显子、内含子、剪接接头和启动子区域中注释的任何人类变体。SInBaD 为启动子、外显子和内含子分别构建了单独的数学模型,使用人类基因突变数据库中注释的人类疾病突变作为功能变体的训练数据集。十折交叉验证显示出较高的预测准确性。在测试数据集上的验证表明,预测为功能的变体在癌症患者中出现的频率显著更高。我们还将我们的模型应用于四个不同个体人类基因组中的变体,以确定一组可能对进一步研究感兴趣的功能变体。所有注释基因的任何可能变体的分数都可在 http://tingchenlab.cmb.usc.edu/sinbad/ 下获得。SInBaD 支持当前的基因分型标准格式,即变体调用文件(VCF 4.0),使其易于集成到任何现有的下一代测序管道中。SNP 检测的准确性是使用 SInBaD 的唯一限制。