Program in Computational Biology and Bioinformatics, Duke University, Durham, North Carolina 27708, USA.
Genome Res. 2012 Sep;22(9):1711-22. doi: 10.1101/gr.135129.111.
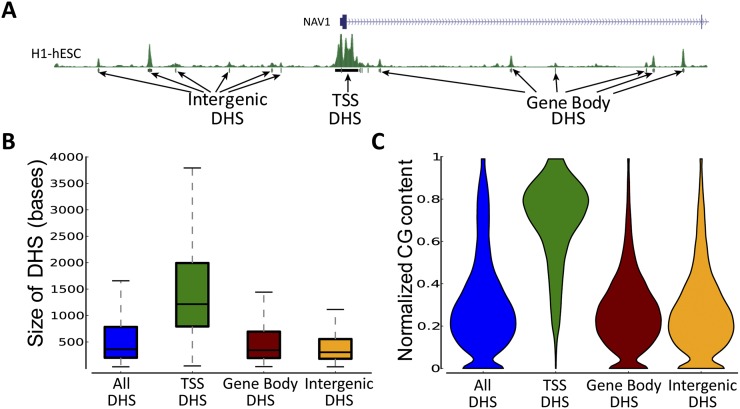
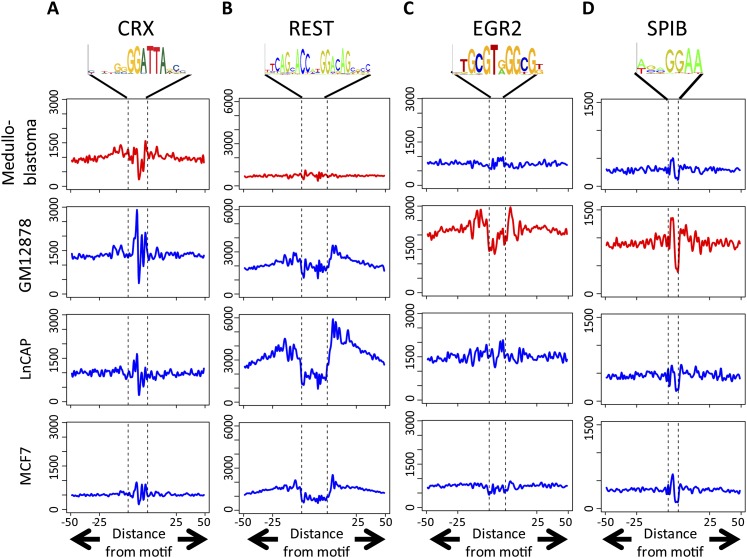
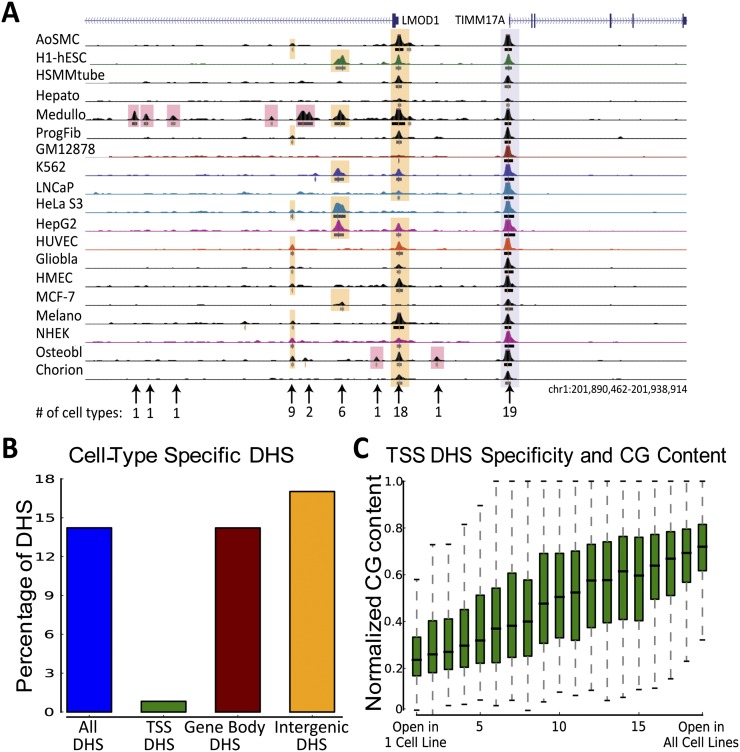
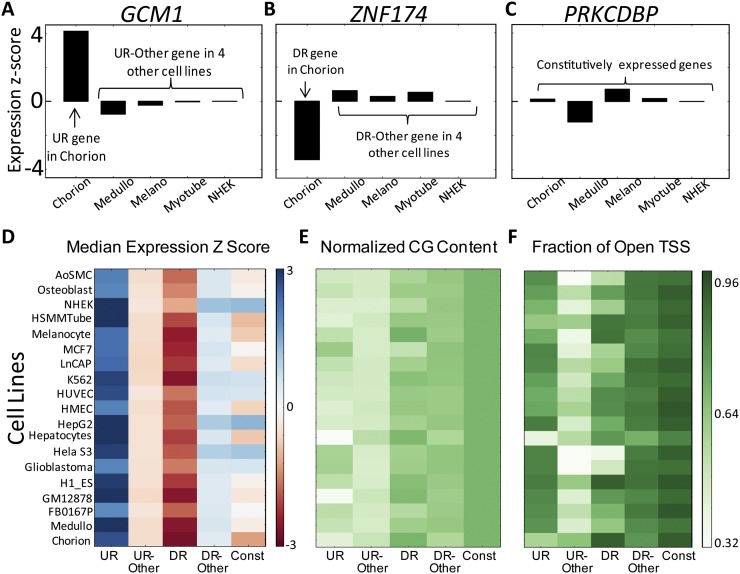
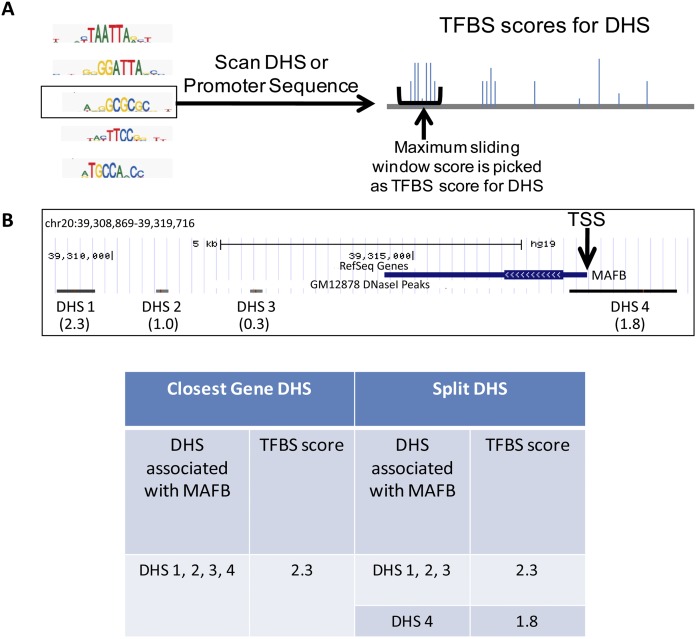
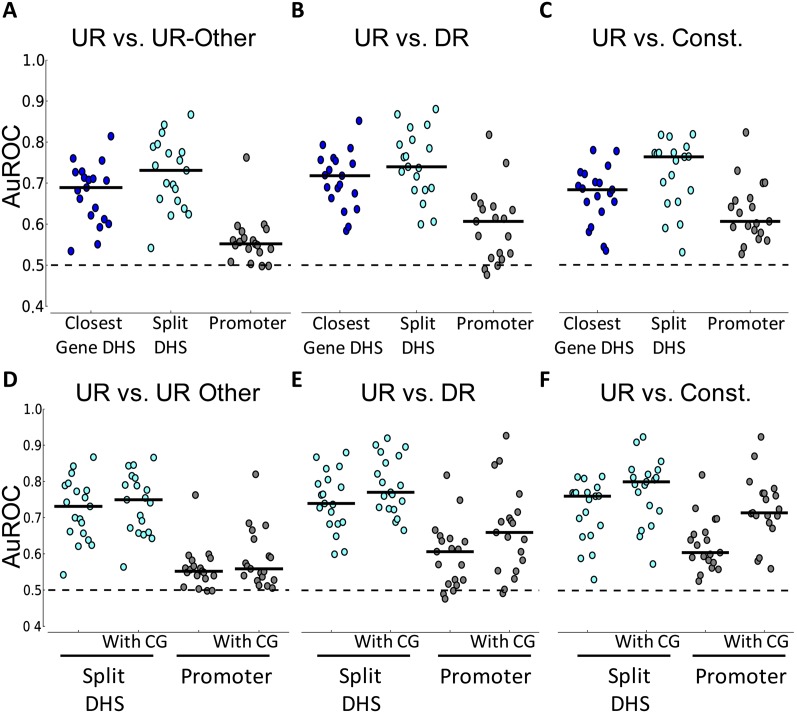
Complex patterns of cell-type-specific gene expression are thought to be achieved by combinatorial binding of transcription factors (TFs) to sequence elements in regulatory regions. Predicting cell-type-specific expression in mammals has been hindered by the oftentimes unknown location of distal regulatory regions. To alleviate this bottleneck, we used DNase-seq data from 19 diverse human cell types to identify proximal and distal regulatory elements at genome-wide scale. Matched expression data allowed us to separate genes into classes of cell-type-specific up-regulated, down-regulated, and constitutively expressed genes. CG dinucleotide content and DNA accessibility in the promoters of these three classes of genes displayed substantial differences, highlighting the importance of including these aspects in modeling gene expression. We associated DNase I hypersensitive sites (DHSs) with genes, and trained classifiers for different expression patterns. TF sequence motif matches in DHSs provided a strong performance improvement in predicting gene expression over the typical baseline approach of using proximal promoter sequences. In particular, we achieved competitive performance when discriminating up-regulated genes from different cell types or genes up- and down-regulated under the same conditions. We identified previously known and new candidate cell-type-specific regulators. The models generated testable predictions of activating or repressive functions of regulators. DNase I footprints for these regulators were indicative of their direct binding to DNA. In summary, we successfully used information of open chromatin obtained by a single assay, DNase-seq, to address the problem of predicting cell-type-specific gene expression in mammalian organisms directly from regulatory sequence.
细胞类型特异性基因表达的复杂模式被认为是通过转录因子(TFs)与调控区域中的序列元件的组合结合来实现的。由于远距离调控区域的位置通常未知,因此预测哺乳动物的细胞类型特异性表达受到了阻碍。为了缓解这一瓶颈,我们使用来自 19 种不同人类细胞类型的 DNase-seq 数据,在全基因组范围内识别近端和远端调控元件。匹配的表达数据使我们能够将基因分为细胞类型特异性上调、下调和组成型表达基因的类别。这三类基因的启动子中的 CG 二核苷酸含量和 DNA 可及性显示出显著差异,突出了在建模基因表达时包含这些方面的重要性。我们将 DNase I 超敏位点(DHSs)与基因相关联,并为不同的表达模式训练分类器。DHSs 中的 TF 序列基序匹配在预测基因表达方面提供了比使用近端启动子序列的典型基线方法更强的性能改进。特别是,当区分不同细胞类型的上调基因或在相同条件下上调和下调的基因时,我们取得了有竞争力的性能。我们确定了先前已知和新的候选细胞类型特异性调节剂。这些模型生成的激活或抑制调节剂功能的测试预测。这些调节剂的 DNase I 足迹表明它们直接与 DNA 结合。总之,我们成功地使用了通过单一测定(DNase-seq)获得的开放染色质信息,直接从调控序列解决了预测哺乳动物生物中细胞类型特异性基因表达的问题。