Institute of Computer Science, University of Göttingen, Goldschmidtstr. 7, Göttingen, 37077, Germany.
BMC Bioinformatics. 2012 Sep 11;13:225. doi: 10.1186/1471-2105-13-225.
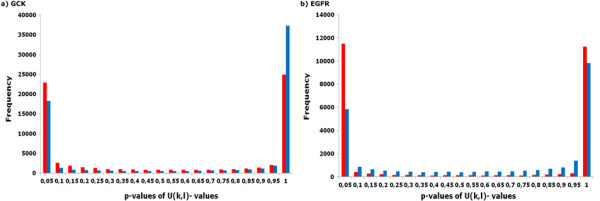
The detection of significant compensatory mutation signals in multiple sequence alignments (MSAs) is often complicated by noise. A challenging problem in bioinformatics is remains the separation of significant signals between two or more non-conserved residue sites from the phylogenetic noise and unrelated pair signals. Determination of these non-conserved residue sites is as important as the recognition of strictly conserved positions for understanding of the structural basis of protein functions and identification of functionally important residue regions. In this study, we developed a new method, the Coupled Mutation Finder (CMF) quantifying the phylogenetic noise for the detection of compensatory mutations.
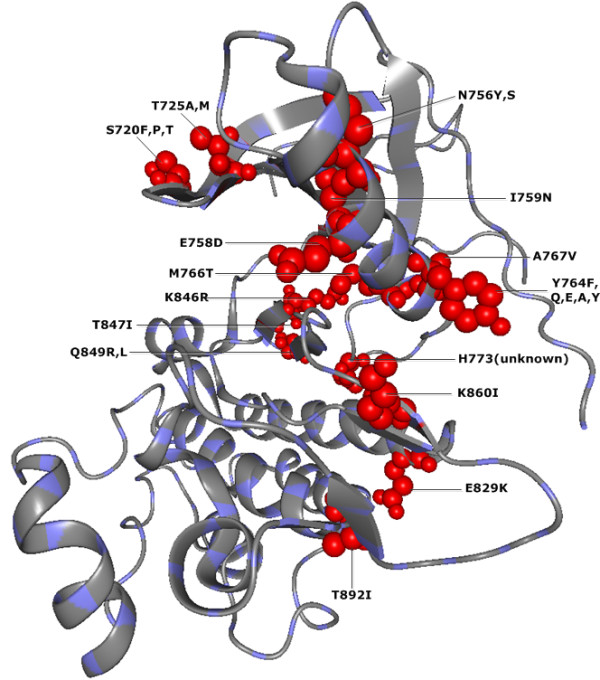
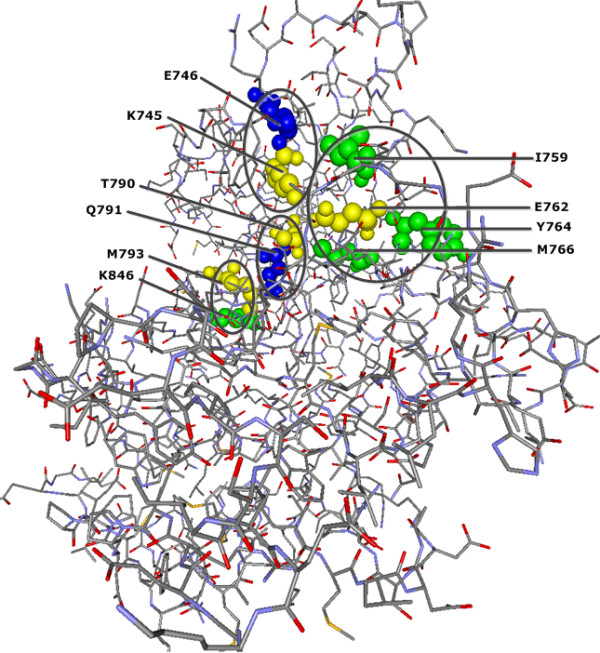
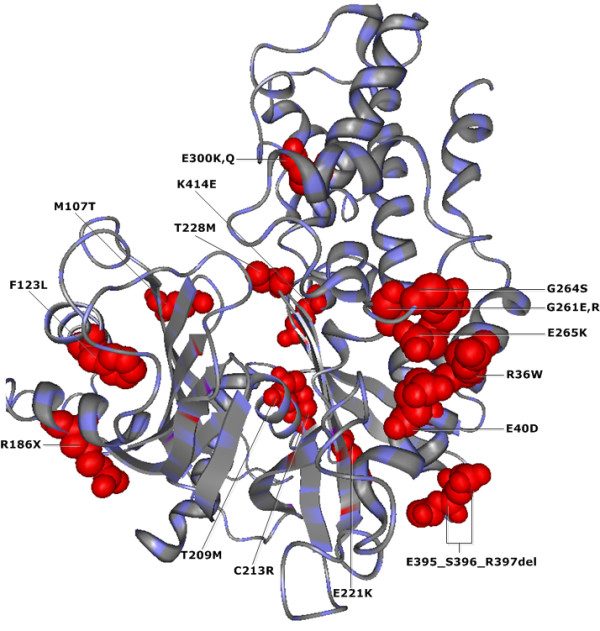
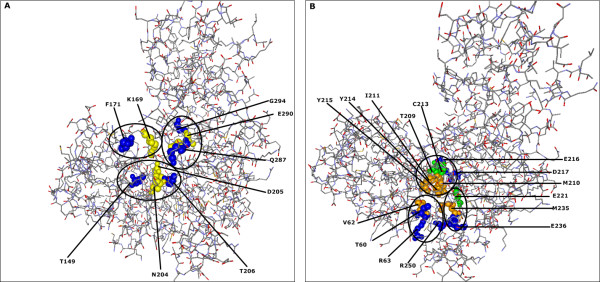
To demonstrate the effectiveness of this method, we analyzed essential sites of two human proteins: epidermal growth factor receptor (EGFR) and glucokinase (GCK). Our results suggest that the CMF is able to separate significant compensatory mutation signals from the phylogenetic noise and unrelated pair signals. The vast majority of compensatory mutation sites found by the CMF are related to essential sites of both proteins and they are likely to affect protein stability or functionality.
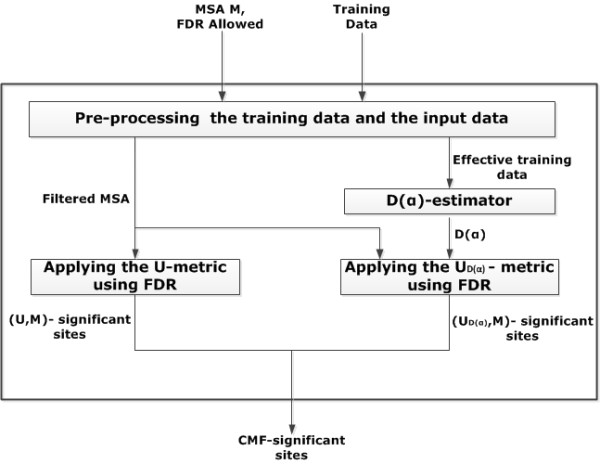
The CMF is a new method, which includes an MSA-specific statistical model based on multiple testing procedures that quantify the error made in terms of the false discovery rate and a novel entropy-based metric to upscale BLOSUM62 dissimilar compensatory mutations. Therefore, it is a helpful tool to predict and investigate compensatory mutation sites of structural or functional importance in proteins. We suggest that the CMF could be used as a novel automated function prediction tool that is required for a better understanding of the structural basis of proteins. The CMF server is freely accessible at http://cmf.bioinf.med.uni-goettingen.de.
在多重序列比对 (MSA) 中检测显著的补偿突变信号通常会受到噪声的干扰。生物信息学中的一个挑战性问题仍然是从系统发育噪声和不相关的对信号中分离两个或多个非保守残基位点之间的显著信号。确定这些非保守残基位点对于理解蛋白质功能的结构基础和识别功能重要的残基区域与识别严格保守的位置同样重要。在这项研究中,我们开发了一种新方法,即耦合突变发现器 (CMF),用于量化检测补偿突变的系统发育噪声。
为了证明该方法的有效性,我们分析了两个人类蛋白质的必需位点:表皮生长因子受体 (EGFR) 和葡萄糖激酶 (GCK)。我们的结果表明,CMF 能够将显著的补偿突变信号与系统发育噪声和不相关的对信号区分开来。CMF 发现的绝大多数补偿突变位点与这两种蛋白质的必需位点有关,它们可能会影响蛋白质的稳定性或功能。
CMF 是一种新方法,它包括一个基于多重测试程序的 MSA 特定的统计模型,该模型以假发现率来量化错误,并使用一种新的基于熵的度量来放大 BLOSUM62 不相似的补偿突变。因此,它是预测和研究蛋白质结构或功能重要性补偿突变位点的有用工具。我们建议将 CMF 用作一种新的自动化功能预测工具,以更好地理解蛋白质的结构基础。CMF 服务器可在 http://cmf.bioinf.med.uni-goettingen.de 免费访问。