Novo Nordisk Foundation Center for Biosustainability, Department of Chemical and Biological Engineering, Chalmers University of Technology, SE-41296, Gothenburg, Sweden.
Nucleic Acids Res. 2012 Nov 1;40(20):10084-97. doi: 10.1093/nar/gks804. Epub 2012 Sep 10.
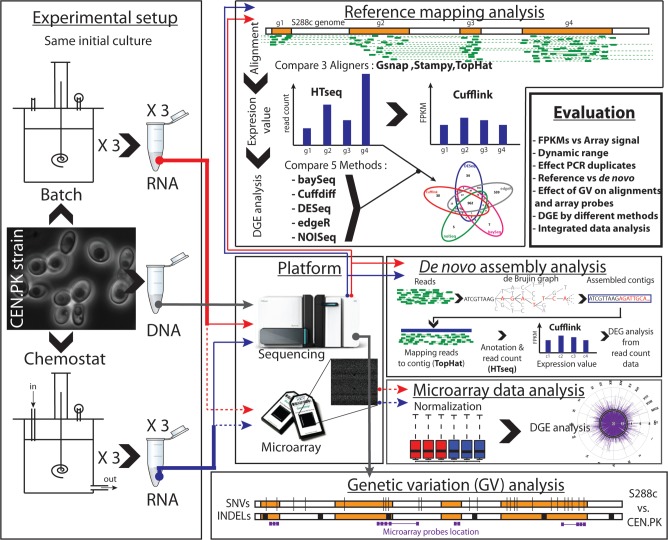
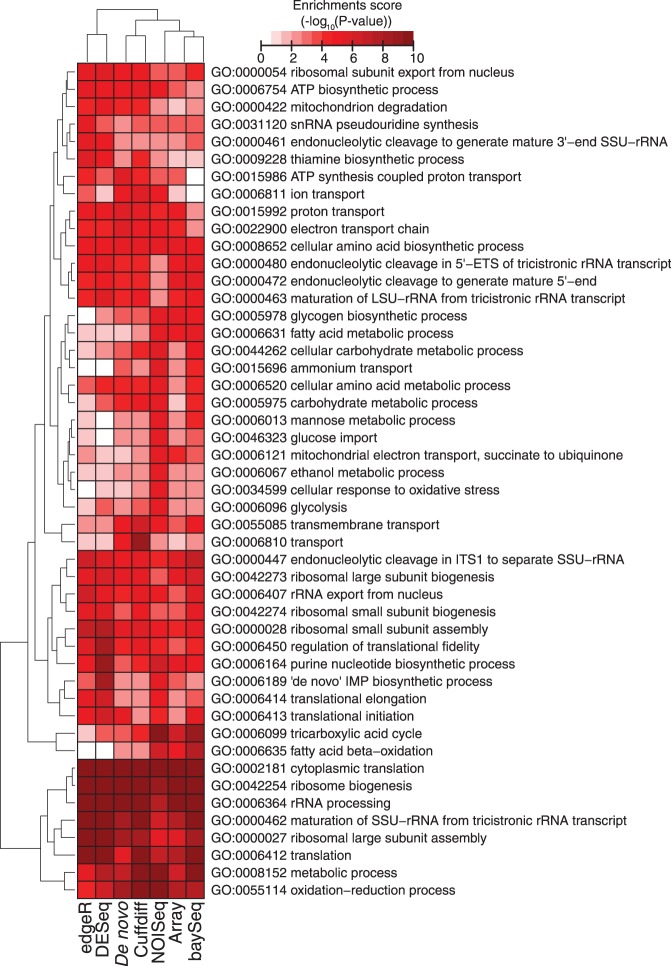
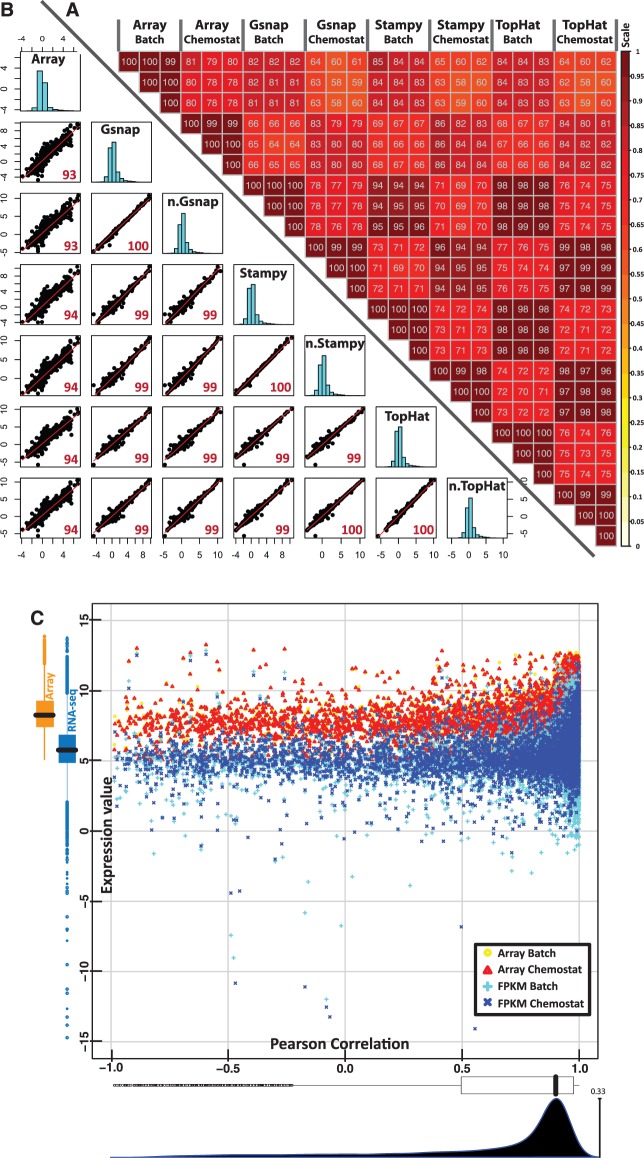
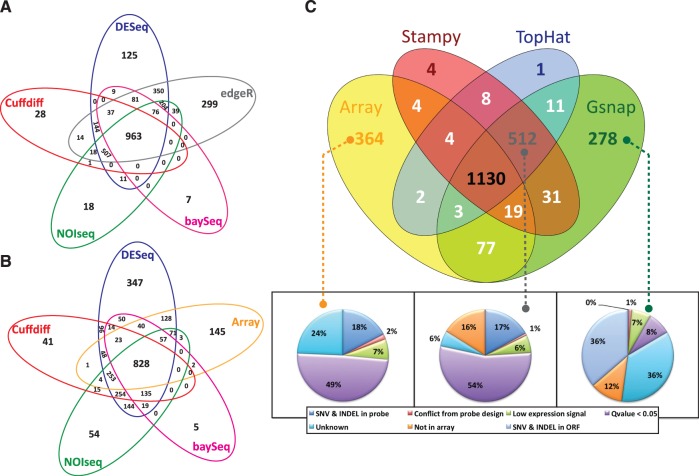
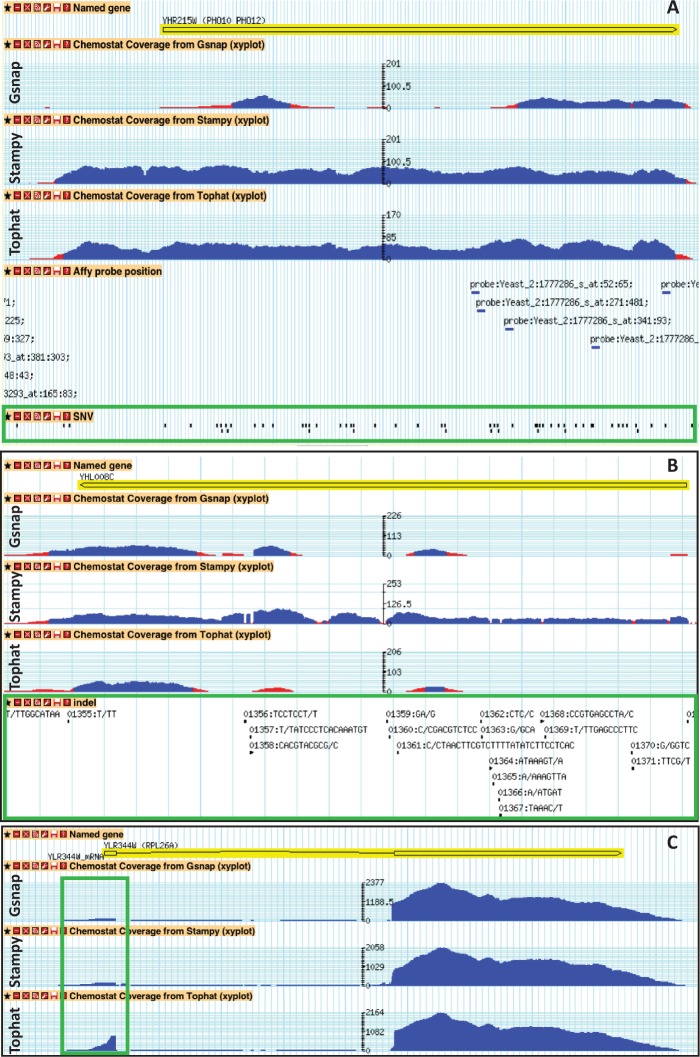
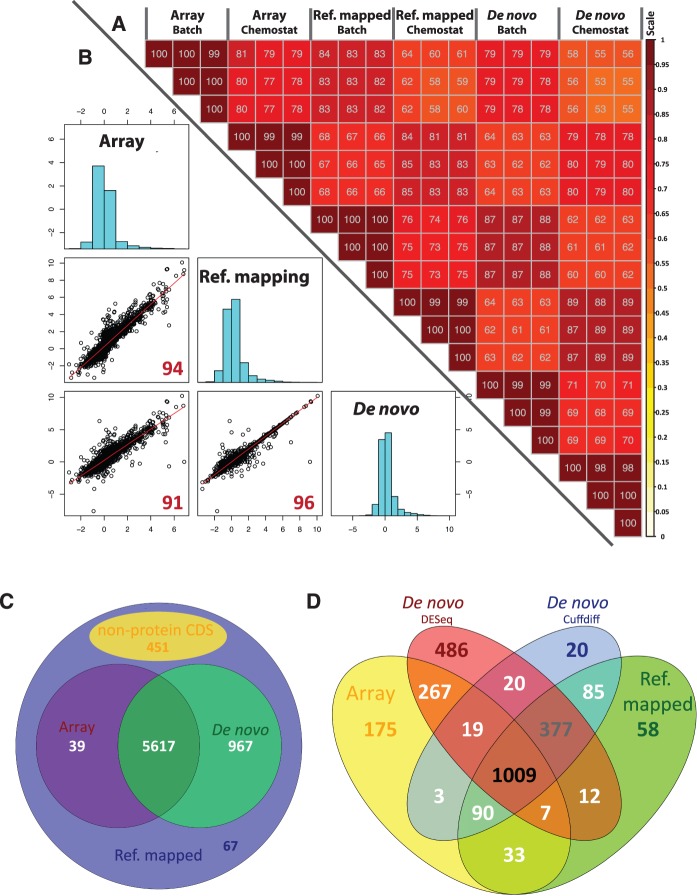
RNA-seq, has recently become an attractive method of choice in the studies of transcriptomes, promising several advantages compared with microarrays. In this study, we sought to assess the contribution of the different analytical steps involved in the analysis of RNA-seq data generated with the Illumina platform, and to perform a cross-platform comparison based on the results obtained through Affymetrix microarray. As a case study for our work we, used the Saccharomyces cerevisiae strain CEN.PK 113-7D, grown under two different conditions (batch and chemostat). Here, we asses the influence of genetic variation on the estimation of gene expression level using three different aligners for read-mapping (Gsnap, Stampy and TopHat) on S288c genome, the capabilities of five different statistical methods to detect differential gene expression (baySeq, Cuffdiff, DESeq, edgeR and NOISeq) and we explored the consistency between RNA-seq analysis using reference genome and de novo assembly approach. High reproducibility among biological replicates (correlation≥0.99) and high consistency between the two platforms for analysis of gene expression levels (correlation≥0.91) are reported. The results from differential gene expression identification derived from the different statistical methods, as well as their integrated analysis results based on gene ontology annotation are in good agreement. Overall, our study provides a useful and comprehensive comparison between the two platforms (RNA-seq and microrrays) for gene expression analysis and addresses the contribution of the different steps involved in the analysis of RNA-seq data.
RNA-seq 最近成为转录组研究中一种极具吸引力的选择方法,与微阵列相比具有多项优势。在这项研究中,我们试图评估 Illumina 平台生成的 RNA-seq 数据分析中涉及的不同分析步骤的贡献,并基于通过 Affymetrix 微阵列获得的结果进行跨平台比较。作为我们工作的案例研究,我们使用了在两种不同条件(分批和恒化器)下生长的酿酒酵母菌株 CEN.PK 113-7D。在这里,我们使用三种不同的读映射比对器(Gsnap、Stampy 和 TopHat)评估遗传变异对基因表达水平估计的影响,对 S288c 基因组进行评估,使用五种不同的统计方法(baySeq、Cuffdiff、DESeq、edgeR 和 NOISeq)来检测差异基因表达,并探索使用参考基因组和从头组装方法进行 RNA-seq 分析的一致性。报告了生物重复之间的高度可重复性(相关性≥0.99)和两种平台之间用于分析基因表达水平的高度一致性(相关性≥0.91)。不同统计方法得出的差异基因表达识别结果以及基于基因本体论注释的综合分析结果非常吻合。总体而言,我们的研究为基因表达分析的两种平台(RNA-seq 和 microrrays)提供了有用且全面的比较,并解决了 RNA-seq 数据分析中涉及的不同步骤的贡献问题。