Department of Biological Sciences, University of Waikato, Hamilton, New Zealand.
PLoS One. 2012;7(9):e44224. doi: 10.1371/journal.pone.0044224. Epub 2012 Sep 6.
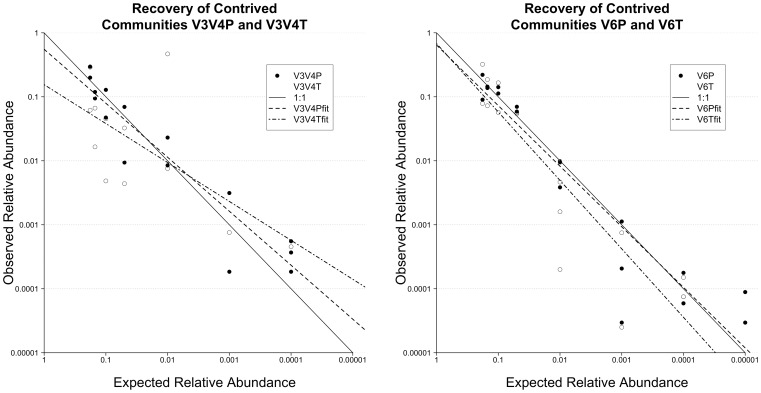
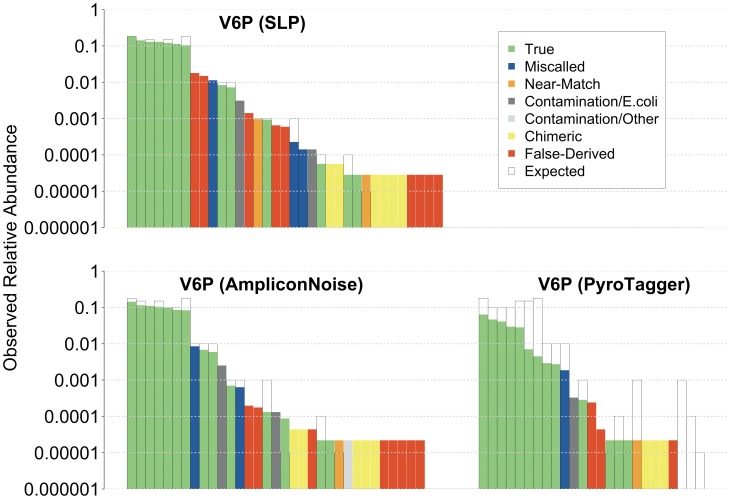
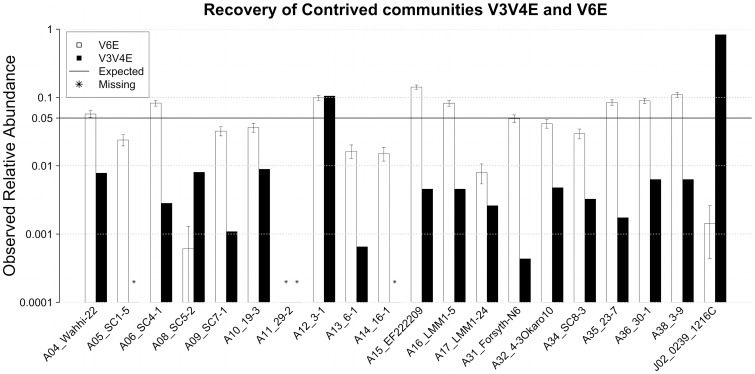
Analysis of microbial communities by high-throughput pyrosequencing of SSU rRNA gene PCR amplicons has transformed microbial ecology research and led to the observation that many communities contain a diverse assortment of rare taxa-a phenomenon termed the Rare Biosphere. Multiple studies have investigated the effect of pyrosequencing read quality on operational taxonomic unit (OTU) richness for contrived communities, yet there is limited information on the fidelity of community structure estimates obtained through this approach. Given that PCR biases are widely recognized, and further unknown biases may arise from the sequencing process itself, a priori assumptions about the neutrality of the data generation process are at best unvalidated. Furthermore, post-sequencing quality control algorithms have not been explicitly evaluated for the accuracy of recovered representative sequences and its impact on downstream analyses, reducing useful discussion on pyrosequencing reads to their diversity and abundances. Here we report on community structures and sequences recovered for in vitro-simulated communities consisting of twenty 16S rRNA gene clones tiered at known proportions. PCR amplicon libraries of the V3-V4 and V6 hypervariable regions from the in vitro-simulated communities were sequenced using the Roche 454 GS FLX Titanium platform. Commonly used quality control protocols resulted in the formation of OTUs with >1% abundance composed entirely of erroneous sequences, while over-aggressive clustering approaches obfuscated real, expected OTUs. The pyrosequencing process itself did not appear to impose significant biases on overall community structure estimates, although the detection limit for rare taxa may be affected by PCR amplicon size and quality control approach employed. Meanwhile, PCR biases associated with the initial amplicon generation may impose greater distortions in the observed community structure.
通过高通量焦磷酸测序 SSU rRNA 基因 PCR 扩增子分析微生物群落已经改变了微生物生态学研究,并导致了这样一种观察结果,即许多群落含有多样化的稀有分类群-这种现象被称为稀有生物圈。多项研究已经调查了 pyrosequencing 读质量对人工群落操作分类单元 (OTU) 丰富度的影响,但是对于通过这种方法获得的群落结构估计的保真度的信息有限。鉴于 PCR 偏倚是广泛公认的,并且可能进一步出现测序过程本身带来的未知偏倚,关于数据生成过程中立性的先验假设充其量是未经证实的。此外,测序后质量控制算法并未明确评估恢复代表性序列的准确性及其对下游分析的影响,这减少了对 pyrosequencing 读的有用讨论,使其多样性和丰度成为焦点。在这里,我们报告了由已知比例的二十个 16S rRNA 基因克隆分层组成的体外模拟群落的群落结构和回收序列。使用 Roche 454 GS FLX Titanium 平台对来自体外模拟群落的 V3-V4 和 V6 高变区的 PCR 扩增子文库进行测序。常用的质量控制协议导致形成了> 1%丰度的 OTUs,其完全由错误序列组成,而过于激进的聚类方法则混淆了真实的、预期的 OTUs。焦磷酸测序过程本身似乎不会对整体群落结构估计产生重大偏差,尽管稀有分类群的检测限可能会受到 PCR 扩增子大小和采用的质量控制方法的影响。与此同时,与初始扩增子生成相关的 PCR 偏倚可能会对观察到的群落结构造成更大的扭曲。