Department of Chemistry, Massachusetts Institute of Technology, 77 Massachusetts Ave., Cambridge, MA 02139, USA.
Metallomics. 2012 Oct;4(10):1020-36. doi: 10.1039/c2mt20142a. Epub 2012 Sep 18.
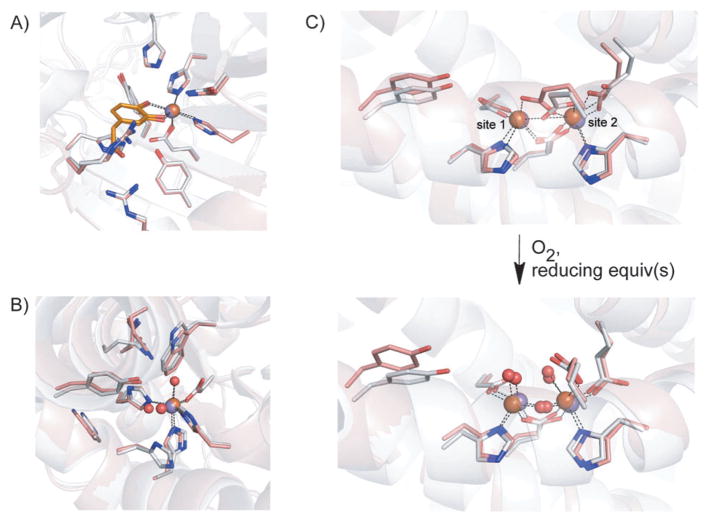
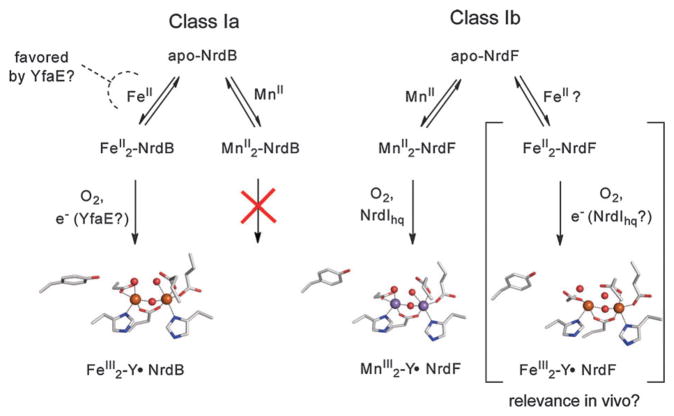
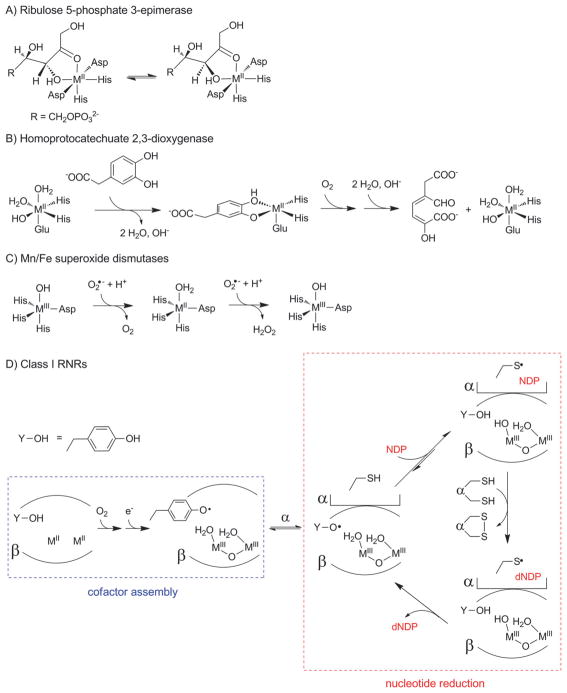
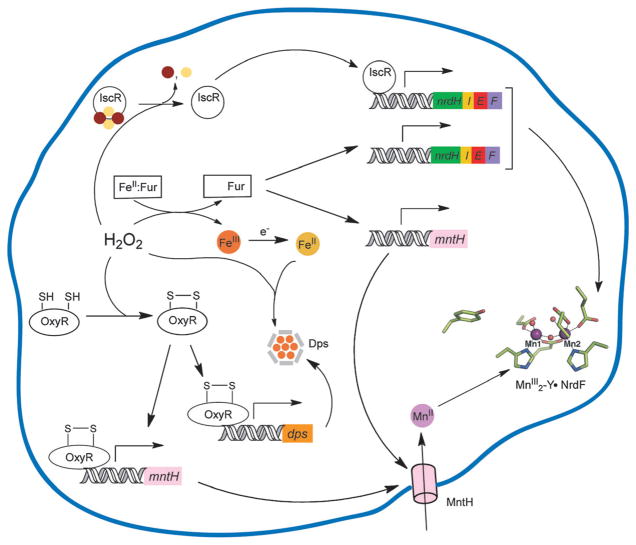
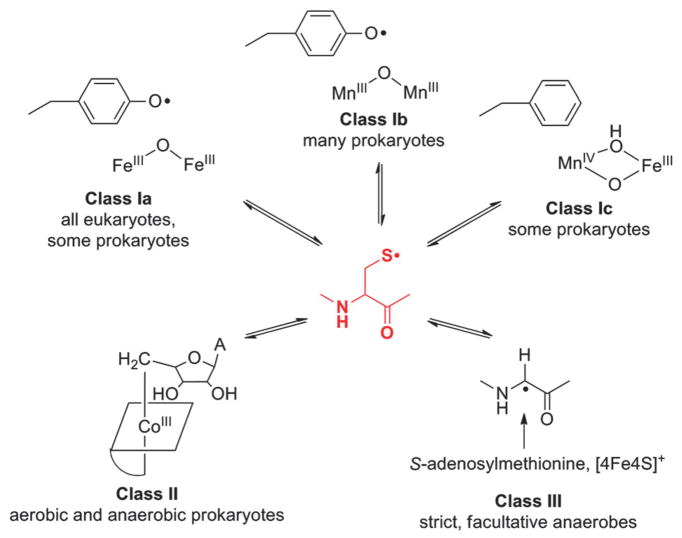
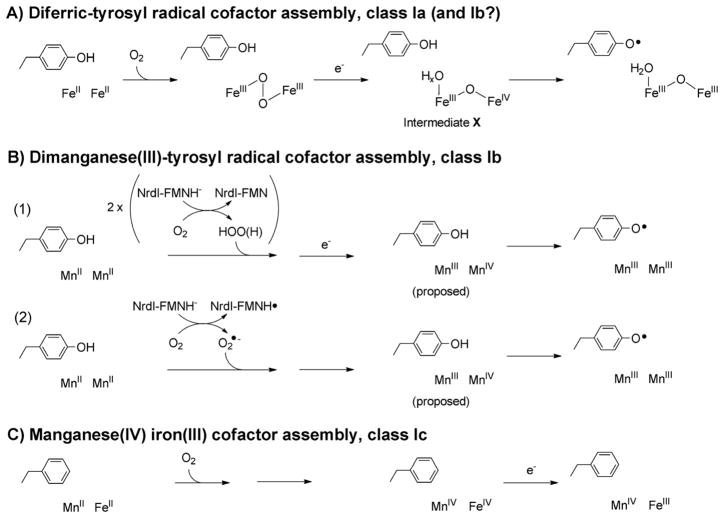
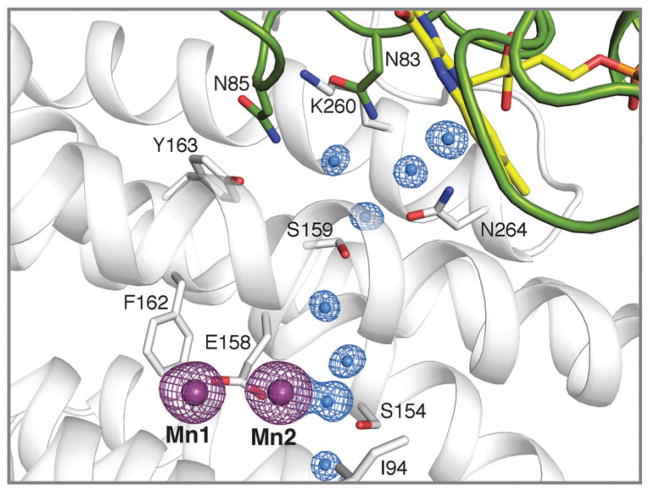
How cells ensure correct metallation of a given protein and whether a degree of promiscuity in metal binding has evolved are largely unanswered questions. In a classic case, iron- and manganese-dependent superoxide dismutases (SODs) catalyze the disproportionation of superoxide using highly similar protein scaffolds and nearly identical active sites. However, most of these enzymes are active with only one metal, although both metals can bind in vitro and in vivo. Iron(ii) and manganese(ii) bind weakly to most proteins and possess similar coordination preferences. Their distinct redox properties suggest that they are unlikely to be interchangeable in biological systems except when they function in Lewis acid catalytic roles, yet recent work suggests this is not always the case. This review summarizes the diversity of ways in which iron and manganese are substituted in similar or identical protein frameworks. As models, we discuss (1) enzymes, such as epimerases, thought to use Fe(II) as a Lewis acid under normal growth conditions but which switch to Mn(II) under oxidative stress; (2) extradiol dioxygenases, which have been found to use both Fe(II) and Mn(II), the redox role of which in catalysis remains to be elucidated; (3) SODs, which use redox chemistry and are generally metal-specific; and (4) the class I ribonucleotide reductases (RNRs), which have evolved unique biosynthetic pathways to control metallation. The primary focus is the class Ib RNRs, which can catalyze formation of a stable radical on a tyrosine residue in their β2 subunits using either a di-iron or a recently characterized dimanganese cofactor. The physiological roles of enzymes that can switch between iron and manganese cofactors are discussed, as are insights obtained from the studies of many groups regarding iron and manganese homeostasis and the divergent and convergent strategies organisms use for control of protein metallation. We propose that, in many of the systems discussed, "discrimination" between metals is not performed by the protein itself, but it is instead determined by the environment in which the protein is expressed.
细胞如何确保给定蛋白质的正确金属化,以及金属结合的“混杂性”是否已经进化,这些在很大程度上仍是尚未解答的问题。在一个经典案例中,铁锰依赖的超氧化物歧化酶(SOD)利用高度相似的蛋白质支架和几乎相同的活性位点来催化超氧化物的歧化反应。然而,这些酶中的大多数仅对一种金属具有活性,尽管这两种金属都可以在体外和体内结合。二价铁(Fe(II))和二价锰(Mn(II))与大多数蛋白质结合较弱,且具有相似的配位偏好。它们不同的氧化还原特性表明,它们在生物系统中不太可能相互替换,除非它们在路易斯酸催化作用中发挥作用,但最近的研究表明情况并非总是如此。本综述总结了在相似或相同的蛋白质框架中替代铁和锰的多样性方式。作为模型,我们讨论了(1)在正常生长条件下被认为使用 Fe(II)作为路易斯酸的酶,例如差向异构酶,但在氧化应激下会切换到 Mn(II);(2)外二醇双加氧酶,已发现其使用 Fe(II)和 Mn(II),其在催化中的氧化还原作用仍有待阐明;(3)SOD,其利用氧化还原化学且通常具有金属特异性;以及(4)I 类核糖核苷酸还原酶(RNR),其已经进化出独特的生物合成途径来控制金属化。主要焦点是 Ib 类 RNR,它可以使用其β2 亚基中的酪氨酸残基上的二价铁或最近表征的二锰辅助因子来催化稳定自由基的形成。讨论了可以在铁和锰辅助因子之间切换的酶的生理作用,以及许多研究小组在铁和锰稳态以及生物体用于控制蛋白质金属化的发散和收敛策略方面获得的见解。我们提出,在讨论的许多系统中,“区分”金属不是由蛋白质本身完成的,而是由蛋白质表达的环境决定的。