Department of Computer Science, Hong Kong Baptist University, Kowloon Tong, Hong Kong, China.
Nucleic Acids Res. 2012 Nov;40(21):10657-67. doi: 10.1093/nar/gks860. Epub 2012 Sep 21.
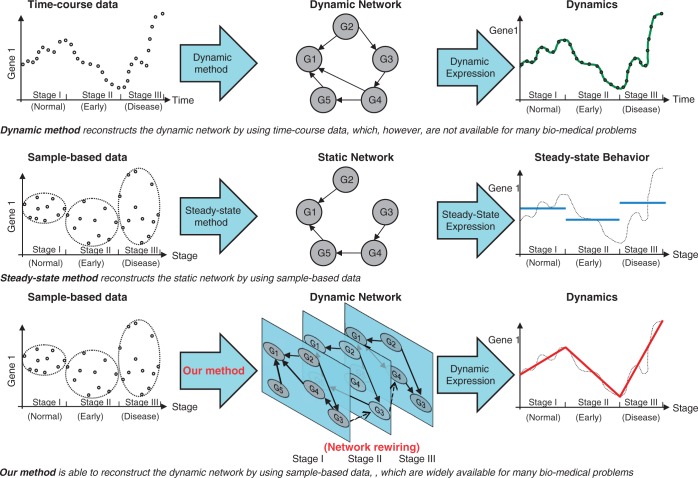
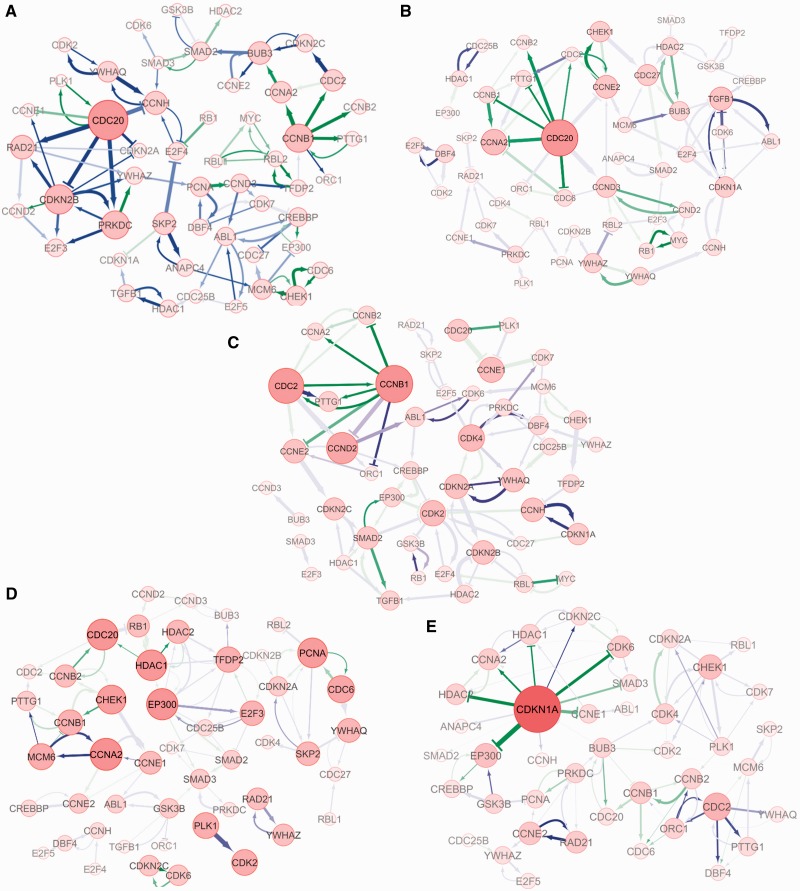
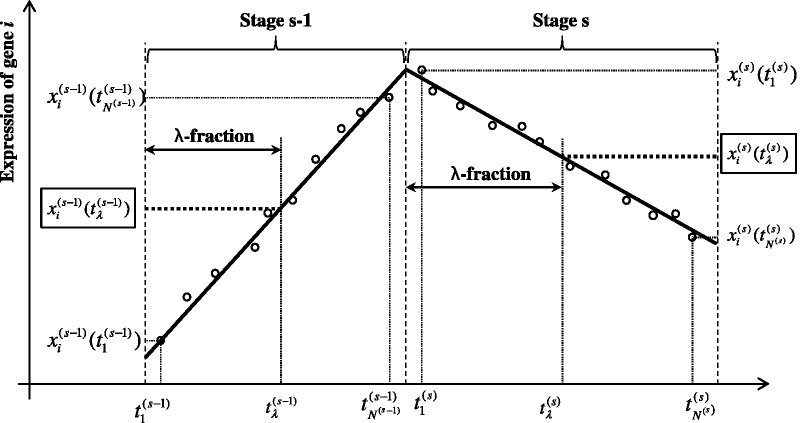
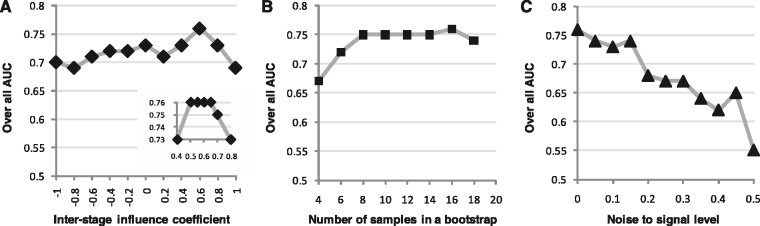
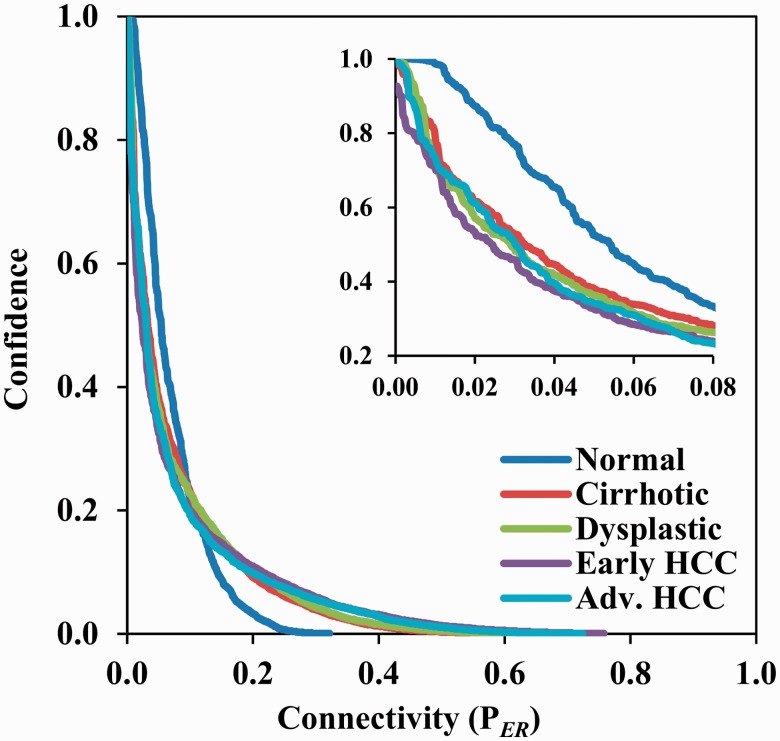
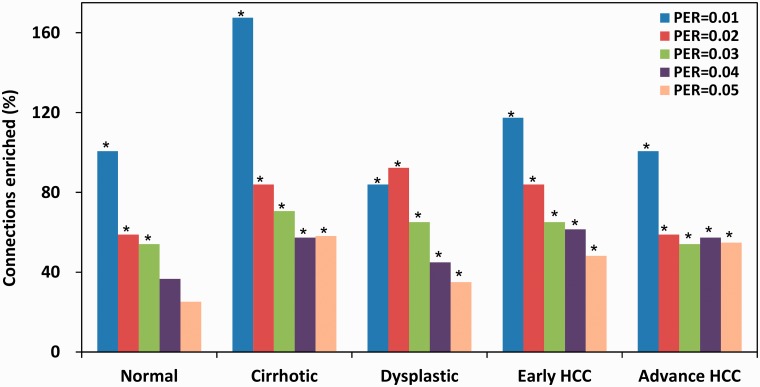
The current method for reconstructing gene regulatory networks faces a dilemma concerning the study of bio-medical problems. On the one hand, static approaches assume that genes are expressed in a steady state and thus cannot exploit and describe the dynamic patterns of an evolving process. On the other hand, approaches that can describe the dynamical behaviours require time-course data, which are normally not available in many bio-medical studies. To overcome the limitations of both the static and dynamic approaches, we propose a dynamic cascaded method (DCM) to reconstruct dynamic gene networks from sample-based transcriptional data. Our method is based on the intra-stage steady-rate assumption and the continuity assumption, which can properly characterize the dynamic and continuous nature of gene transcription in a biological process. Our simulation study showed that compared with static approaches, the DCM not only can reconstruct dynamical network but also can significantly improve network inference performance. We further applied our method to reconstruct the dynamic gene networks of hepatocellular carcinoma (HCC) progression. The derived HCC networks were verified by functional analysis and network enrichment analysis. Furthermore, it was shown that the modularity and network rewiring in the HCC networks can clearly characterize the dynamic patterns of HCC progression.
当前的基因调控网络重建方法在研究生物医学问题时面临着一个困境。一方面,静态方法假设基因在稳态下表达,因此无法利用和描述不断演变的过程中的动态模式。另一方面,能够描述动态行为的方法需要时程数据,但在许多生物医学研究中通常无法获得。为了克服静态和动态方法的局限性,我们提出了一种动态级联方法(DCM),用于从基于样本的转录数据中重建动态基因网络。我们的方法基于阶段内稳态率假设和连续性假设,这可以正确描述生物过程中基因转录的动态和连续性。我们的模拟研究表明,与静态方法相比,DCM 不仅可以重建动态网络,还可以显著提高网络推断性能。我们进一步将我们的方法应用于重建肝细胞癌(HCC)进展的动态基因网络。通过功能分析和网络富集分析验证了所得到的 HCC 网络。此外,结果表明 HCC 网络中的模块性和网络重布线可以清楚地描述 HCC 进展的动态模式。