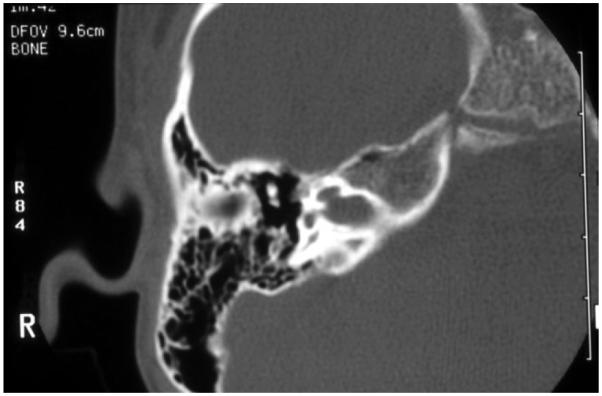
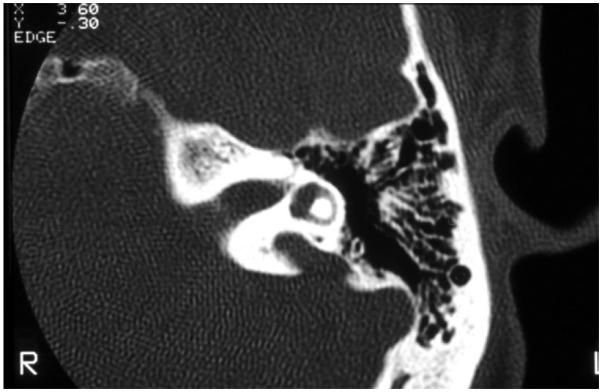
Department of Otolaryngology, University of Miami, Miami, Florida 33136, USA.
Anat Rec (Hoboken). 2012 Nov;295(11):1812-29. doi: 10.1002/ar.22579. Epub 2012 Oct 8.
This article is a review of the genes and genetic disorders that affect hearing in humans and a few selected mouse models of deafness. Genetics is playing an increasingly critical role in the practice of medicine. This is not only in part to the importance that genetic knowledge has on traditional genetic diseases but also in part to the fact that genetic knowledge provides an understanding of the fundamental biological process of most diseases. The proteins coded by the genes related to hearing loss (HL) are involved in many functions in the ear, such as cochlear fluid homeostasis, ionic channels, stereocilia morphology and function, synaptic transmission, gene regulation, and others. Mouse models play a crucial role in understanding of the pathogenesis associated with these genes. Different types of familial HL have been recognized for years; however, in the last two decades, there has been tremendous progress in the discovery of gene mutations that cause deafness. Most of the cases of genetic deafness recognized today are monogenic disorders that can be broadly classified by the mode of inheritance (i.e., autosomal dominant, autosomal recessive, X-linked, and mitochondrial inheritance) and by the presence of associated phenotypic features (i.e., syndromic; and nonsyndromic). In terms of nonsyndromic HL, the chromosomal locations are currently known for ∼ 125 loci (54 for dominant and 71 for recessive deafness), 64 genes have been identified (24 for dominant and 40 for recessive deafness), and there are many more loci for syndromic deafness and X-linked and mitochondrial DNA disorders (http://hereditaryhearingloss.org). Thus, today's clinician must understand the science of medical genetics as this knowledge can lead to more effective disease diagnosis, counseling, treatment, and prevention.
这是一篇关于影响人类听力的基因和遗传疾病的综述,以及一些选择的耳聋小鼠模型。遗传学在医学实践中发挥着越来越关键的作用。这不仅部分归因于遗传知识对传统遗传疾病的重要性,还部分归因于遗传知识提供了对大多数疾病基本生物学过程的理解。与听力损失(HL)相关的基因编码的蛋白质参与耳朵的许多功能,例如耳蜗液的动态平衡、离子通道、静纤毛形态和功能、突触传递、基因调控等。小鼠模型在理解与这些基因相关的发病机制方面起着至关重要的作用。多年来已经认识到不同类型的家族性 HL;然而,在过去的二十年中,发现导致耳聋的基因突变取得了巨大进展。今天公认的大多数遗传性耳聋都是单基因疾病,可以根据遗传方式(即常染色体显性、常染色体隐性、X 连锁和线粒体遗传)和相关表型特征(即综合征性和非综合征性)进行广泛分类。在非综合征性 HL 方面,目前已知约 125 个基因座(显性 54 个,隐性 71 个)的染色体位置,已鉴定出 64 个基因(显性 24 个,隐性 40 个),还有更多的综合征性耳聋和 X 连锁及线粒体 DNA 疾病的基因座(http://hereditaryhearingloss.org)。因此,今天的临床医生必须了解医学遗传学的科学,因为这一知识可以导致更有效的疾病诊断、咨询、治疗和预防。