Alere Technologies GmbH, Jena, Germany.
PLoS One. 2012;7(10):e46489. doi: 10.1371/journal.pone.0046489. Epub 2012 Oct 4.
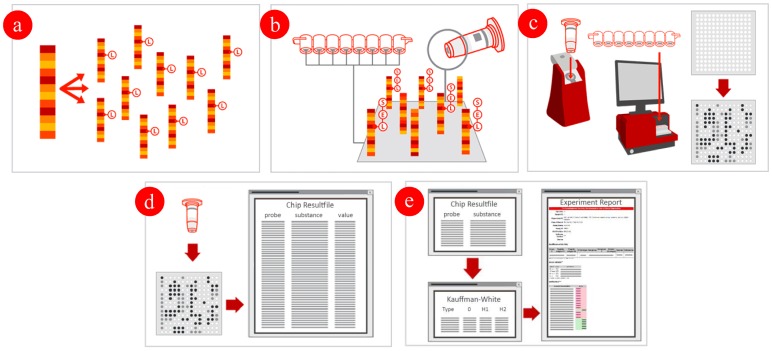
Salmonellosis caused by Salmonella (S.) belongs to the most prevalent food-borne zoonotic diseases throughout the world. Therefore, serotype identification for all culture-confirmed cases of Salmonella infection is important for epidemiological purposes. As a standard, the traditional culture method (ISO 6579:2002) is used to identify Salmonella. Classical serotyping takes 4-5 days to be completed, it is labor-intensive, expensive and more than 250 non-standardized sera are necessary to characterize more than 2,500 Salmonella serovars currently known. These technical difficulties could be overcome with modern molecular methods. We developed a microarray based serogenotyping assay for the most prevalent Salmonella serovars in Europe and North America. The current assay version could theoretically discriminate 28 O-antigens and 86 H-antigens. Additionally, we included 77 targets analyzing antimicrobial resistance genes. The Salmonella assay was evaluated with a set of 168 reference strains representing 132 serovars previously serotyped by conventional agglutination through various reference centers. 117 of 132 (81%) tested serovars showed an unique microarray pattern. 15 of 132 serovars generated a pattern which was shared by multiple serovars (e.g., S. ser. Enteritidis and S. ser. Nitra). These shared patterns mainly resulted from the high similarity of the genotypes of serogroup A and D1. Using patterns of the known reference strains, a database was build which represents the basis of a new PatternMatch software that can serotype unknown Salmonella isolates automatically. After assay verification, the Salmonella serogenotyping assay was used to identify a field panel of 105 Salmonella isolates. All were identified as Salmonella and 93 of 105 isolates (88.6%) were typed in full concordance with conventional serotyping. This microarray based assay is a powerful tool for serogenotyping.
由沙门氏菌(S.)引起的沙门氏菌病属于全世界最普遍的食源性人畜共患病。因此,对所有经培养确认的沙门氏菌感染病例进行血清型鉴定对于流行病学目的非常重要。作为标准,传统的培养方法(ISO 6579:2002)用于鉴定沙门氏菌。经典的血清分型需要 4-5 天才能完成,它是劳动密集型的,昂贵的,并且需要超过 250 种非标准化血清来描述目前已知的超过 2500 种沙门氏菌血清型。这些技术困难可以通过现代分子方法克服。我们开发了一种基于微阵列的欧洲和北美的最常见沙门氏菌血清型的血清分型分析方法。当前的分析版本理论上可以区分 28 个 O 抗原和 86 个 H 抗原。此外,我们还包括了 77 个分析抗生素耐药基因的目标。沙门氏菌分析方法用一组 168 个参考菌株进行了评估,这些参考菌株代表了 132 个以前通过各种参考中心进行传统凝集法血清分型的血清型。132 个测试血清型中的 117 个(81%)显示出独特的微阵列模式。132 个血清型中的 15 个产生了多个血清型共享的模式(例如,S. ser. Enteritidis 和 S. ser. Nitra)。这些共享模式主要是由于 A 组和 D1 组的基因型高度相似所致。使用已知参考菌株的模式,构建了一个数据库,该数据库是一种新的 PatternMatch 软件的基础,该软件可以自动对未知的沙门氏菌分离株进行血清型分析。在对分析方法进行验证后,该沙门氏菌血清分型分析方法用于鉴定了 105 个沙门氏菌分离株的现场面板。所有鉴定均为沙门氏菌,105 个分离株中有 93 个(88.6%)与传统血清分型完全一致。这种基于微阵列的分析方法是一种强大的血清型分析工具。