Carter Robert W, Sanford John C
FMS Foundation, 877 Marshall Rd, Waterloo, NY 13165, USA.
Theor Biol Med Model. 2012 Oct 12;9:42. doi: 10.1186/1742-4682-9-42.
The H1N1 influenza A virus has been circulating in the human population for over 95 years, first manifesting itself in the pandemic of 1917-1918. Initial mortality was extremely high, but dropped exponentially over time. Influenza viruses have high mutation rates, and H1N1 has undergone significant genetic changes since 1918. The exact nature of H1N1 mutation accumulation over time has not been fully explored.
We have made a comprehensive historical analysis of mutational changes within H1N1 by examining over 4100 fully-sequenced H1N1 genomes. This has allowed us to examine the genetic changes arising within H1N1 from 1918 to the present.
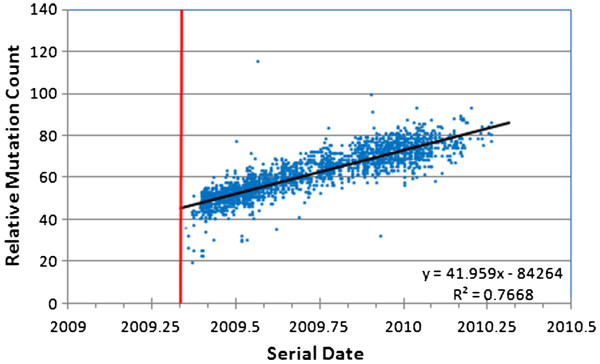
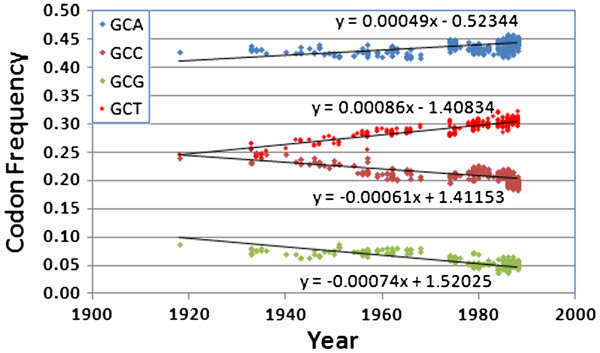
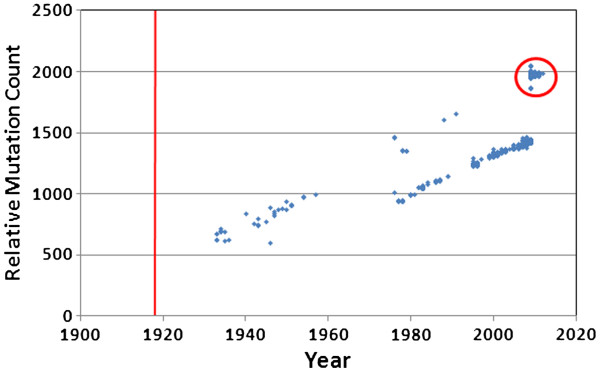
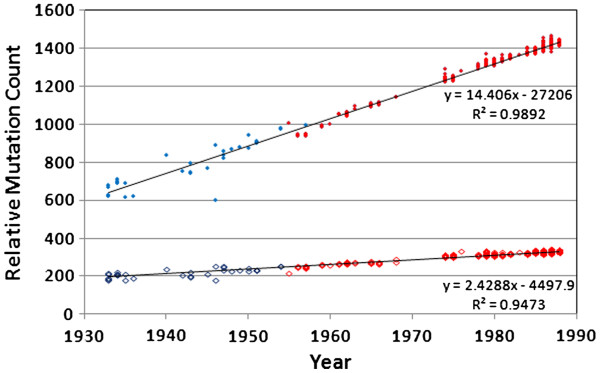
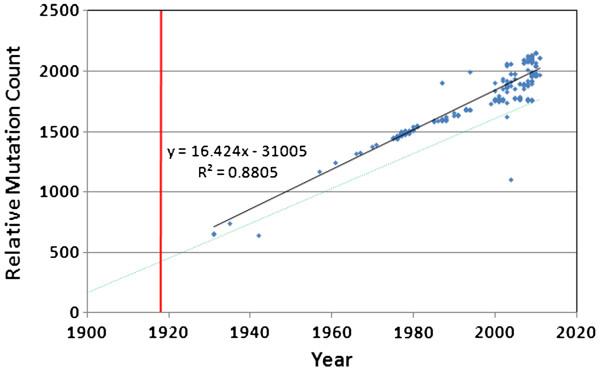
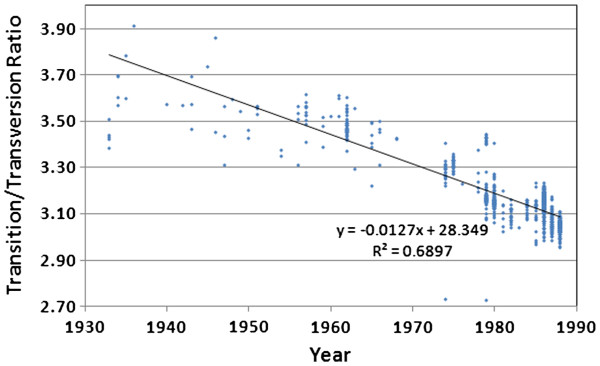
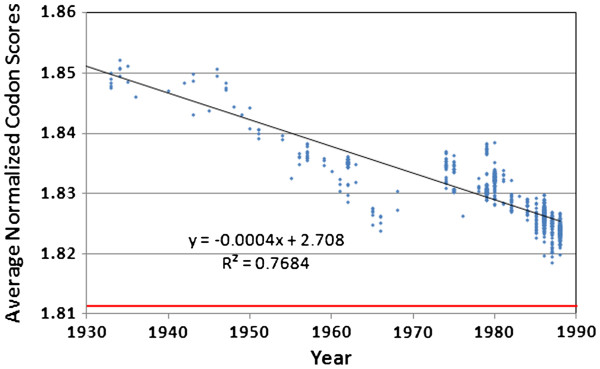
We document multiple extinction events, including the previously known extinction of the human H1N1 lineage in the 1950s, and an apparent second extinction of the human H1N1 lineage in 2009. These extinctions appear to be due to a continuous accumulation of mutations. At the time of its disappearance in 2009, the human H1N1 lineage had accumulated over 1400 point mutations (more than 10% of the genome), including approximately 330 non-synonymous changes (7.4% of all codons). The accumulation of both point mutations and non-synonymous amino acid changes occurred at constant rates (μ = 14.4 and 2.4 new mutations/year, respectively), and mutations accumulated uniformly across the entire influenza genome. We observed a continuous erosion over time of codon-specificity in H1N1, including a shift away from host (human, swine, and bird [duck]) codon preference patterns.
While there have been numerous adaptations within the H1N1 genome, most of the genetic changes we document here appear to be non-adaptive, and much of the change appears to be degenerative. We suggest H1N1 has been undergoing natural genetic attenuation, and that significant attenuation may even occur during a single pandemic. This process may play a role in natural pandemic cessation and has apparently contributed to the exponential decline in mortality rates over time, as seen in all major human influenza strains. These findings may be relevant to the development of strategies for managing influenza pandemics and strain evolution.
甲型H1N1流感病毒已在人群中传播了95年以上,首次出现在1917 - 1918年的大流行中。最初的死亡率极高,但随着时间的推移呈指数下降。流感病毒具有高突变率,自1918年以来H1N1经历了重大的基因变化。H1N1随时间积累突变的确切性质尚未得到充分探索。
我们通过检查4100多个全测序的H1N1基因组,对H1N1内的突变变化进行了全面的历史分析。这使我们能够研究1918年至现在H1N1内发生的基因变化。
我们记录了多次灭绝事件,包括20世纪50年代人类H1N1谱系先前已知的灭绝,以及2009年人类H1N1谱系明显的第二次灭绝。这些灭绝似乎是由于突变的持续积累。在2009年消失时,人类H1N1谱系已经积累了超过1400个点突变(超过基因组的10%),包括大约330个非同义变化(占所有密码子的7.4%)。点突变和非同义氨基酸变化的积累都以恒定速率发生(分别为μ = 14.4和2.4个新突变/年),并且突变在整个流感基因组中均匀积累。我们观察到H1N1中密码子特异性随时间持续受到侵蚀,包括偏离宿主(人类、猪和鸟类[鸭])密码子偏好模式。
虽然H1N1基因组内有许多适应性变化,但我们在此记录的大多数基因变化似乎是非适应性的,而且许多变化似乎是退化性的。我们认为H1N1一直在经历自然基因衰减,甚至在一次大流行期间可能会发生显著衰减。这一过程可能在自然大流行停止中起作用,并且显然导致了随着时间推移死亡率的指数下降,这在所有主要人类流感毒株中都有体现。这些发现可能与制定管理流感大流行和毒株进化的策略相关。