Turku Centre for Biotechnology, University of Turku and Åbo Akademi University, Tykistökatu 6, 20520, Turku, Finland.
BMC Genomics. 2012 Oct 30;13:575. doi: 10.1186/1471-2164-13-575.
Lichens are symbiotic organisms that have a remarkable ability to survive in some of the most extreme terrestrial climates on earth. Lichens can endure frequent desiccation and wetting cycles and are able to survive in a dehydrated molecular dormant state for decades at a time. Genetic resources have been established in lichen species for the study of molecular systematics and their taxonomic classification. No lichen species have been characterised yet using genomics and the molecular mechanisms underlying the lichen symbiosis and the fundamentals of desiccation tolerance remain undescribed. We report the characterisation of a transcriptome of the grey reindeer lichen, Cladonia rangiferina, using high-throughput next-generation transcriptome sequencing and traditional Sanger EST sequencing data.
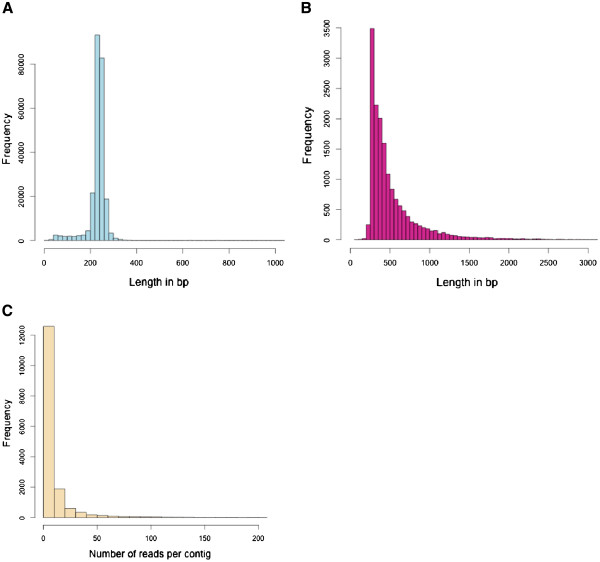
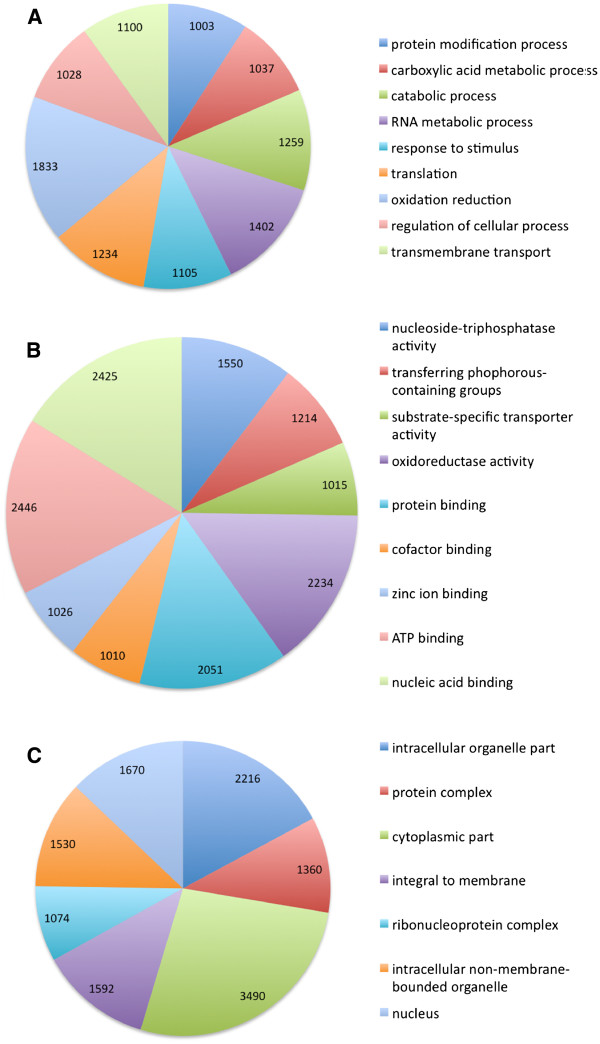
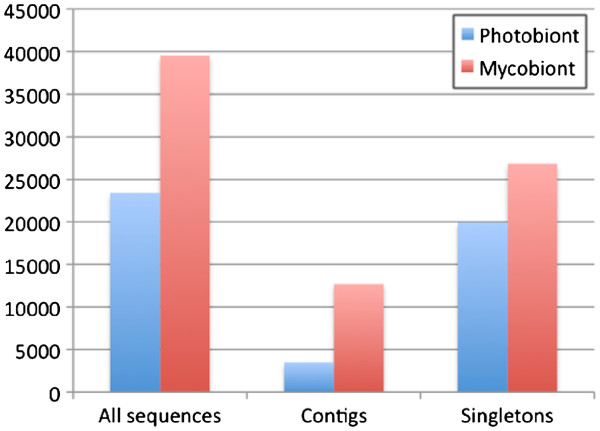
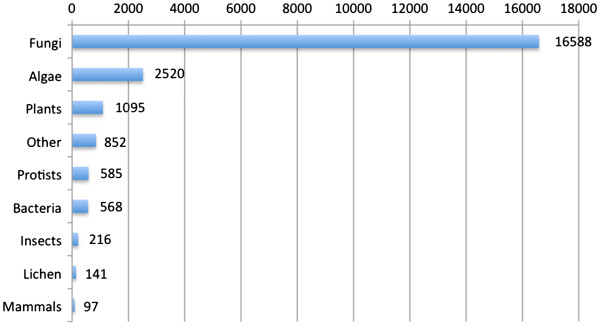
Altogether 243,729 high quality sequence reads were de novo assembled into 16,204 contigs and 49,587 singletons. The genome of origin for the sequences produced was predicted using Eclat with sequences derived from the axenically grown symbiotic partners used as training sequences for the classification model. 62.8% of the sequences were classified as being of fungal origin while the remaining 37.2% were predicted as being of algal origin. The assembled sequences were annotated by BLASTX comparison against a non-redundant protein sequence database with 34.4% of the sequences having a BLAST match. 29.3% of the sequences had a Gene Ontology term match and 27.9% of the sequences had a domain or structural match following an InterPro search. 60 KEGG pathways with more than 10 associated sequences were identified.
Our results present a first transcriptome sequencing and de novo assembly for a lichen species and describe the ongoing molecular processes and the most active pathways in C. rangiferina. This brings a meaningful contribution to publicly available lichen sequence information. These data provide a first glimpse into the molecular nature of the lichen symbiosis and characterise the transcriptional space of this remarkable organism. These data will also enable further studies aimed at deciphering the genetic mechanisms behind lichen desiccation tolerance.
地衣是共生生物,具有在地球上一些最极端的陆地气候中生存的非凡能力。地衣可以耐受频繁的干燥和润湿循环,并且能够在脱水的分子休眠状态下一次存活数十年。遗传资源已在地衣物种中建立,用于研究分子系统发育及其分类学分类。尚未使用基因组学对地衣物种进行特征描述,地衣共生的分子机制和干燥耐受性的基本原理仍未被描述。我们报告了使用高通量下一代转录组测序和传统 Sanger EST 测序数据对地衣物种灰色驯鹿地衣 Cladonia rangiferina 的转录组进行的特征描述。
总共将 243,729 条高质量序列从头组装成 16,204 个 contigs 和 49,587 个 singletons。使用 Eclat 预测产生的序列的基因组起源,使用共生的共生伙伴的体外培养物序列作为分类模型的训练序列。62.8%的序列被分类为真菌起源,而其余 37.2%被预测为藻类起源。组装的序列通过 BLASTX 与非冗余蛋白质序列数据库进行比较进行注释,其中 34.4%的序列具有 BLAST 匹配。29.3%的序列具有基因本体术语匹配,27.9%的序列在进行 InterPro 搜索后具有域或结构匹配。确定了 60 个具有超过 10 个相关序列的 KEGG 途径。
我们的结果首次对地衣物种进行了转录组测序和从头组装,并描述了 C. rangiferina 中正在进行的分子过程和最活跃的途径。这为公共可用的地衣序列信息做出了有意义的贡献。这些数据首次揭示了地衣共生的分子本质,并描绘了这个非凡生物的转录空间。这些数据还将为进一步研究地衣干燥耐受性背后的遗传机制提供支持。