Department of Biomedical Informatics, Vanderbilt University School of Medicine, Nashville, TN, USA.
BMC Genomics. 2012;13 Suppl 6(Suppl 6):S15. doi: 10.1186/1471-2164-13-S6-S15. Epub 2012 Oct 26.
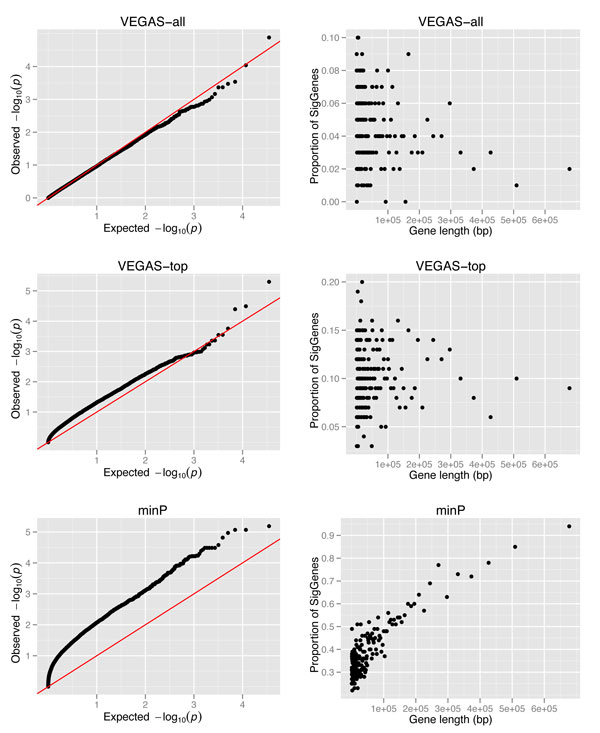
Genome-wide association studies (GWAS) have generated a wealth of valuable genotyping data for complex diseases/traits. A large proportion of these data are embedded with many weakly associated markers that have been missed in traditional single marker analyses, but they may provide valuable insights in dissecting the genetic components of diseases. Gene set analysis (GSA) augmented by protein-protein interaction network data provides a promising way to examine GWAS data by analyzing the combined effects of multiple genes/markers, each of which may have only individually weak to moderate association effects. A critical issue in GSA of GWAS data is the definition of gene-wise P values based on multiple SNPs mapped to a gene.
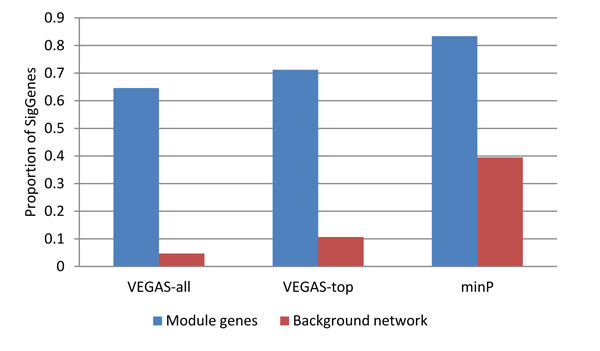
In this study, we proposed an alternative restricted search approach based on our previously developed dense module search algorithm, and we demonstrated it in the CATIE GWAS dataset for schizophrenia. Specifically, we explored three ways of computing gene-wise P values and examined their effects on the resultant module genes. These methods calculate gene-wise P values based on all the SNPs, the top ranked SNPs, or the most significant SNP among all the SNPs mapped to a gene. We applied the restricted search approach and identified a module gene set for each of the gene-wise P value data set. In our evaluation using an independent method, ALIGATOR, we showed that although each of these input datasets generated a unique set of module genes, all of them were significant in the GWAS dataset. Further functional enrichment analysis of these module genes showed that at the pathway level, they were all consistently related to neuro- and immune-related pathways. Finally, we compared our method with a previously reported method.
Our results showed that the approaches to computing gene-wise P values in GWAS data are critical in GSA. This work is useful for evaluating key factors in GSA of GWAS data.
全基因组关联研究 (GWAS) 为复杂疾病/性状生成了大量有价值的基因分型数据。这些数据中有很大一部分包含许多在传统的单标记分析中被忽略的弱相关标记,但它们可能为剖析疾病的遗传成分提供有价值的见解。通过分析多个基因/标记的联合效应,基于蛋白质-蛋白质相互作用网络数据的基因集分析 (GSA) 为检查 GWAS 数据提供了一种很有前途的方法,而每个基因/标记的关联效应可能只有微弱到中等强度。GWAS 数据 GSA 的一个关键问题是基于映射到一个基因的多个 SNP 定义基因水平的 P 值。
在这项研究中,我们基于之前开发的密集模块搜索算法提出了一种替代的受限搜索方法,并在精神分裂症的 CATIE GWAS 数据集上进行了演示。具体来说,我们探索了三种计算基因水平 P 值的方法,并研究了它们对所得模块基因的影响。这些方法基于所有 SNP、排名最高的 SNP 或映射到一个基因的所有 SNP 中最显著的 SNP 计算基因水平的 P 值。我们应用受限搜索方法并为每个基因水平 P 值数据集确定了一个模块基因集。在使用独立方法 ALIGATOR 进行的评估中,我们表明,尽管这些输入数据集中的每一个都生成了一组独特的模块基因,但它们在 GWAS 数据集中都是显著的。对这些模块基因的进一步功能富集分析表明,在通路水平上,它们都与神经和免疫相关通路有关。最后,我们将我们的方法与之前报道的方法进行了比较。
我们的结果表明,计算 GWAS 数据中基因水平 P 值的方法在 GSA 中是至关重要的。这项工作对于评估 GWAS 数据 GSA 的关键因素很有用。