Department of Pharmacology, University of Oxford, Oxford OX1 3QT, England, UK.
J Cell Biol. 2012 Nov 26;199(5):723-34. doi: 10.1083/jcb.201208152.
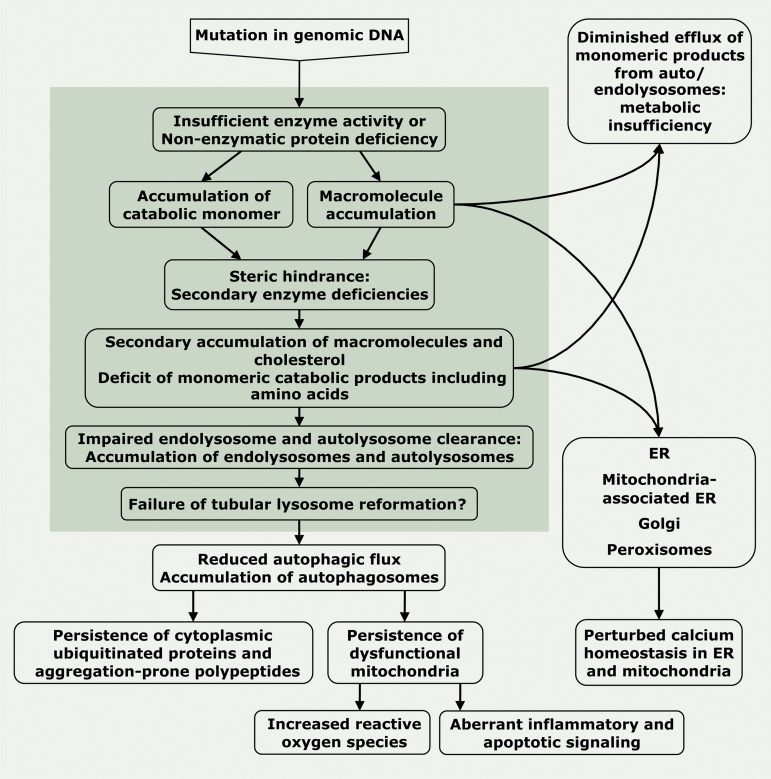
Lysosomal storage diseases (LSDs) are a family of disorders that result from inherited gene mutations that perturb lysosomal homeostasis. LSDs mainly stem from deficiencies in lysosomal enzymes, but also in some non-enzymatic lysosomal proteins, which lead to abnormal storage of macromolecular substrates. Valuable insights into lysosome functions have emerged from research into these diseases. In addition to primary lysosomal dysfunction, cellular pathways associated with other membrane-bound organelles are perturbed in these disorders. Through selective examples, we illustrate why the term "cellular storage disorders" may be a more appropriate description of these diseases and discuss therapies that can alleviate storage and restore normal cellular function.
溶酶体贮积症(LSDs)是一组由遗传基因突变引起的疾病,这些基因突变会破坏溶酶体的内稳态。LSDs 主要源于溶酶体酶的缺乏,但也有一些非酶性溶酶体蛋白的缺乏,这会导致大分子底物的异常储存。对这些疾病的研究为我们深入了解溶酶体功能提供了有价值的线索。除了原发性溶酶体功能障碍外,这些疾病还会干扰与其他膜结合细胞器相关的细胞通路。通过有选择性的例子,我们说明了为什么“细胞贮积症”可能是对这些疾病更恰当的描述,并讨论了可以减轻贮积和恢复正常细胞功能的治疗方法。