Department of Internal Medicine, University of Iowa, Iowa City, IA 52242, USA.
Trends Neurosci. 2011 Aug;34(8):401-10. doi: 10.1016/j.tins.2011.05.006. Epub 2011 Jun 30.
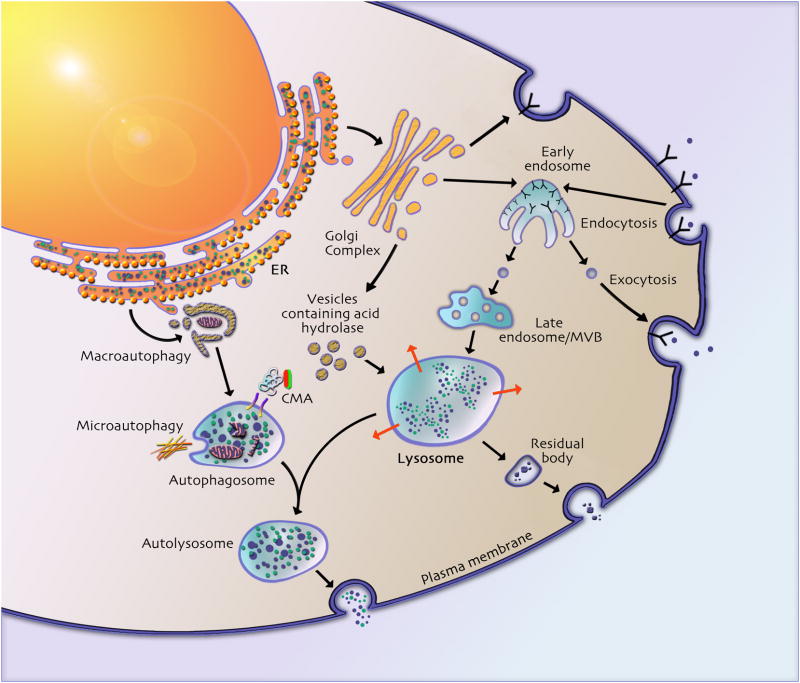
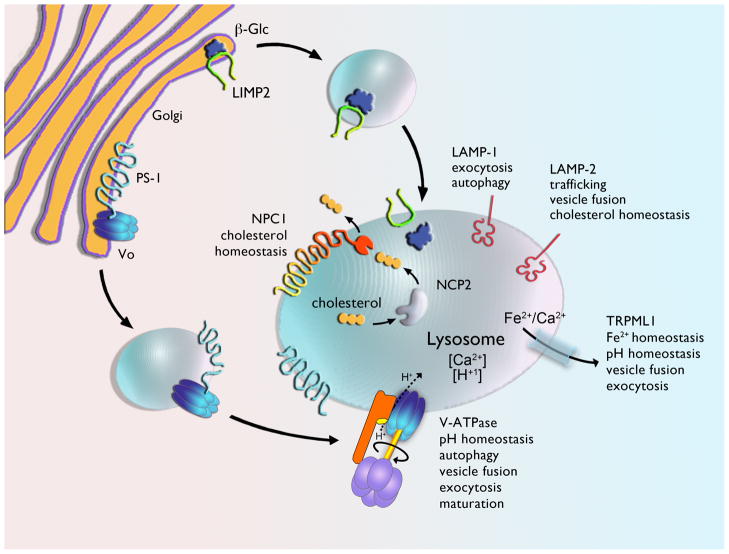
Lysosomal storage diseases (LSDs) are a class of metabolic disorders caused by mutations in proteins critical for lysosomal function. Such proteins include lysosomal enzymes, lysosomal integral membrane proteins, and proteins involved in the post-translational modification and trafficking of lysosomal proteins. There are many recognized forms of LSDs and, although individually rare, their combined prevalence is estimated to be 1 in 8000 births. Over two-thirds of LSDs involve central nervous system (CNS) dysfunction (progressive cognitive and motor decline) and these symptoms are often the most debilitating. Although the genetic basis for these disorders is clear and the biochemistry of the proteins well understood, the cellular mechanisms by which deficiencies in these proteins disrupt neuronal viability remain ambiguous. In this review, we provide an overview of the widespread cellular perturbations occurring in LSDs, how they might be linked and interventions that may specifically or globally correct those defects.
溶酶体贮积症(LSDs)是一类由溶酶体功能关键蛋白的突变引起的代谢性疾病。这些蛋白包括溶酶体酶、溶酶体整合膜蛋白以及参与溶酶体蛋白翻译后修饰和运输的蛋白。有许多公认的 LSD 形式,尽管每种形式都很少见,但它们的总患病率估计为每 8000 例出生中有 1 例。超过三分之二的 LSD 涉及中枢神经系统(CNS)功能障碍(进行性认知和运动功能衰退),这些症状通常是最使人衰弱的。尽管这些疾病的遗传基础很明确,蛋白的生物化学也很清楚,但这些蛋白的缺乏如何破坏神经元活力的细胞机制仍不清楚。在这篇综述中,我们概述了 LSD 中广泛存在的细胞扰动,它们可能如何相关联,以及哪些干预措施可能专门或全局纠正这些缺陷。