School of Medical and Molecular Sciences, and the Ithree Institute at the University of Technology Sydney-UTS, New South Wales, Australia.
PLoS One. 2012;7(11):e50609. doi: 10.1371/journal.pone.0050609. Epub 2012 Nov 30.
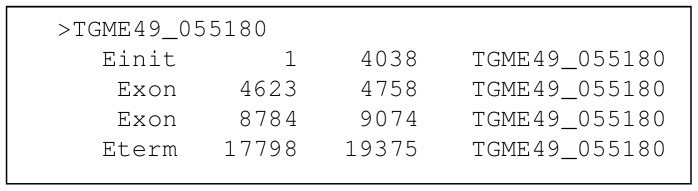
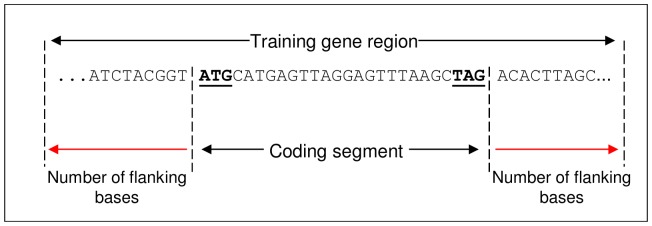
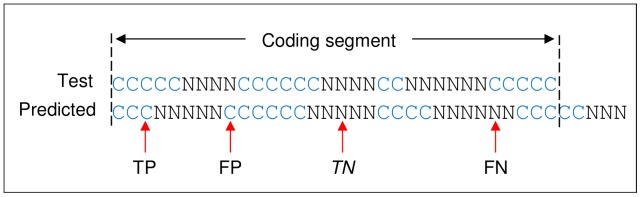
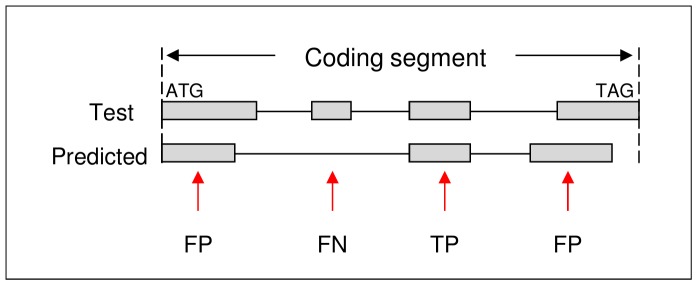
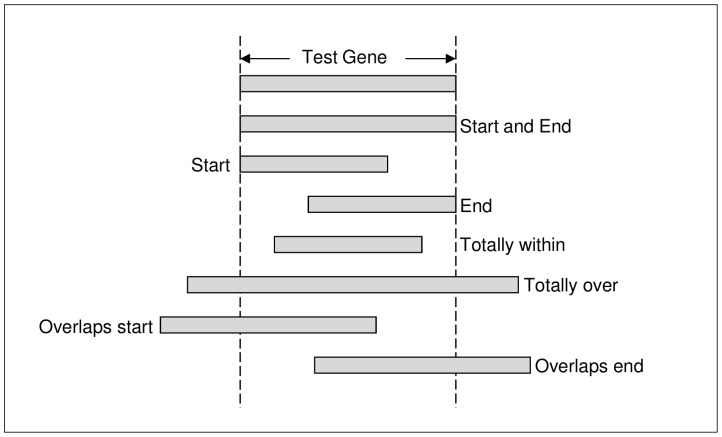
Next generation sequencing technology is advancing genome sequencing at an unprecedented level. By unravelling the code within a pathogen's genome, every possible protein (prior to post-translational modifications) can theoretically be discovered, irrespective of life cycle stages and environmental stimuli. Now more than ever there is a great need for high-throughput ab initio gene finding. Ab initio gene finders use statistical models to predict genes and their exon-intron structures from the genome sequence alone. This paper evaluates whether existing ab initio gene finders can effectively predict genes to deduce proteins that have presently missed capture by laboratory techniques. An aim here is to identify possible patterns of prediction inaccuracies for gene finders as a whole irrespective of the target pathogen. All currently available ab initio gene finders are considered in the evaluation but only four fulfil high-throughput capability: AUGUSTUS, GeneMark_hmm, GlimmerHMM, and SNAP. These gene finders require training data specific to a target pathogen and consequently the evaluation results are inextricably linked to the availability and quality of the data. The pathogen, Toxoplasma gondii, is used to illustrate the evaluation methods. The results support current opinion that predicted exons by ab initio gene finders are inaccurate in the absence of experimental evidence. However, the results reveal some patterns of inaccuracy that are common to all gene finders and these inaccuracies may provide a focus area for future gene finder developers.
下一代测序技术正在以前所未有的水平推进基因组测序。通过揭示病原体基因组内的密码,理论上可以发现每一种可能的蛋白质(在翻译后修饰之前),无论生命周期阶段和环境刺激如何。现在比以往任何时候都更需要高通量的从头基因发现。从头基因发现者使用统计模型仅从基因组序列预测基因及其外显子-内含子结构。本文评估了现有的从头基因发现者是否可以有效地预测基因,以推断目前未被实验室技术捕获的蛋白质。其目的是确定基因发现者整体预测不准确的可能模式,而不考虑目标病原体。在评估中考虑了所有现有的从头基因发现者,但只有四个具有高通量能力:AUGUSTUS、GeneMark_hmm、GlimmerHMM 和 SNAP。这些基因发现者需要针对特定目标病原体的训练数据,因此评估结果与数据的可用性和质量密不可分。寄生虫刚地弓形虫被用来举例说明评估方法。结果支持当前的观点,即在缺乏实验证据的情况下,从头基因发现者预测的外显子是不准确的。然而,结果揭示了所有基因发现者都存在的一些常见的不准确模式,这些不准确模式可能为未来的基因发现者开发者提供一个关注领域。