Dybas Joseph M, Madrid-Aliste Carlos J, Che Fa-Yun, Nieves Edward, Rykunov Dmitry, Angeletti Ruth Hogue, Weiss Louis M, Kim Kami, Fiser Andras
Biodefense Proteomics Research Center, Albert Einstein College of Medicine, Bronx, New York, United States of America.
PLoS One. 2008;3(12):e3899. doi: 10.1371/journal.pone.0003899. Epub 2008 Dec 9.
Toxoplasma gondii is an obligate intracellular protozoan that infects 20 to 90% of the population. It can cause both acute and chronic infections, many of which are asymptomatic, and, in immunocompromised hosts, can cause fatal infection due to reactivation from an asymptomatic chronic infection. An essential step towards understanding molecular mechanisms controlling transitions between the various life stages and identifying candidate drug targets is to accurately characterize the T. gondii proteome.
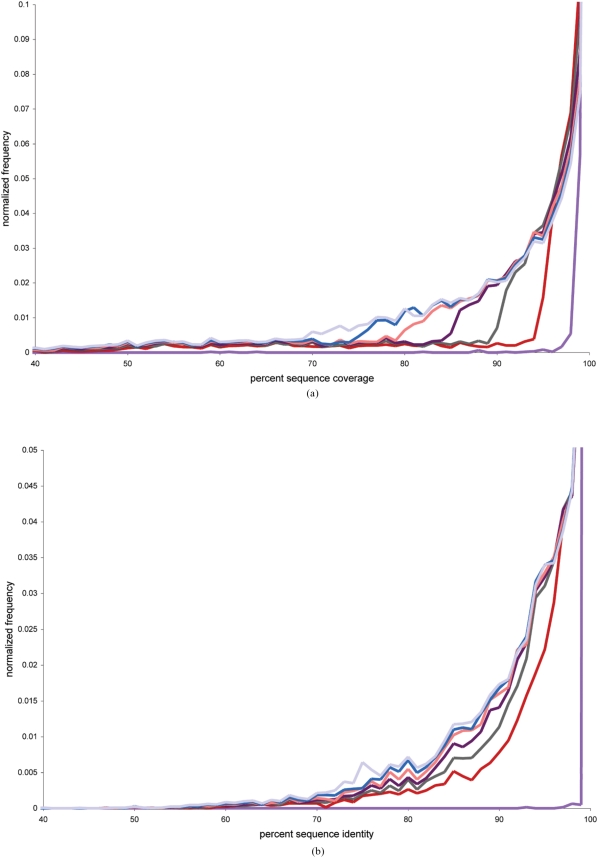
METHODOLOGY/PRINCIPAL FINDINGS: We have explored the proteome of T. gondii tachyzoites with high throughput proteomics experiments and by comparison to publicly available cDNA sequence data. Mass spectrometry analysis validated 2,477 gene coding regions with 6,438 possible alternative gene predictions; approximately one third of the T. gondii proteome. The proteomics survey identified 609 proteins that are unique to Toxoplasma as compared to any known species including other Apicomplexan. Computational analysis identified 787 cases of possible gene duplication events and located at least 6,089 gene coding regions. Commonly used gene prediction algorithms produce very disparate sets of protein sequences, with pairwise overlaps ranging from 1.4% to 12%. Through this experimental and computational exercise we benchmarked gene prediction methods and observed false negative rates of 31 to 43%.
CONCLUSIONS/SIGNIFICANCE: This study not only provides the largest proteomics exploration of the T. gondii proteome, but illustrates how high throughput proteomics experiments can elucidate correct gene structures in genomes.
刚地弓形虫是一种专性细胞内原生动物,感染人群比例为20%至90%。它可引起急性和慢性感染,其中许多感染无症状,并且在免疫功能低下的宿主中,可因无症状慢性感染的激活而导致致命感染。准确表征刚地弓形虫蛋白质组是理解控制不同生命阶段转换的分子机制以及确定候选药物靶点的关键一步。
方法/主要发现:我们通过高通量蛋白质组学实验并与公开可用的cDNA序列数据进行比较,探索了刚地弓形虫速殖子的蛋白质组。质谱分析验证了2477个基因编码区域,有6438个可能的替代基因预测;约占刚地弓形虫蛋白质组的三分之一。蛋白质组学调查确定了609种与任何已知物种(包括其他顶复门动物)相比,刚地弓形虫特有的蛋白质。计算分析确定了787例可能的基因重复事件,并定位了至少6089个基因编码区域。常用的基因预测算法产生的蛋白质序列集差异很大,成对重叠范围从1.4%到12%。通过这项实验和计算工作,我们对基因预测方法进行了基准测试,并观察到假阴性率为31%至43%。
结论/意义:本研究不仅提供了对刚地弓形虫蛋白质组的最大规模蛋白质组学探索,还说明了高通量蛋白质组学实验如何阐明基因组中的正确基因结构。