Center for Health Informatics and Bioinformatics, New York University Langone Medical Center, New York, NY 10016, USA.
BMC Genomics. 2012;13 Suppl 8(Suppl 8):S22. doi: 10.1186/1471-2164-13-S8-S22. Epub 2012 Dec 17.
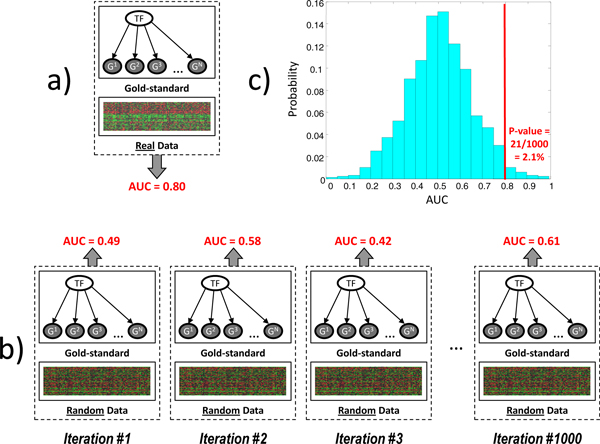
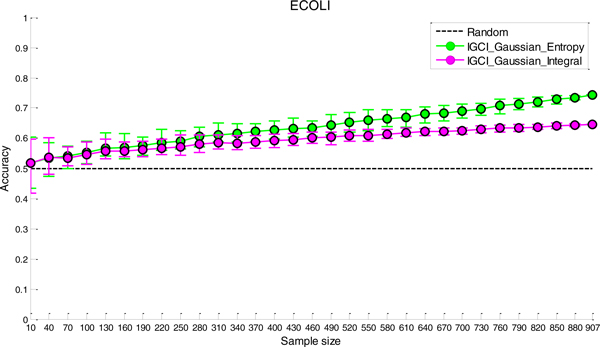
The discovery of molecular pathways is a challenging problem and its solution relies on the identification of causal molecular interactions in genomics data. Causal molecular interactions can be discovered using randomized experiments; however such experiments are often costly, infeasible, or unethical. Fortunately, algorithms that infer causal interactions from observational data have been in development for decades, predominantly in the quantitative sciences, and many of them have recently been applied to genomics data. While these algorithms can infer unoriented causal interactions between involved molecular variables (i.e., without specifying which one is the cause and which one is the effect), causally orienting all inferred molecular interactions was assumed to be an unsolvable problem until recently. In this work, we use transcription factor-target gene regulatory interactions in three different organisms to evaluate a new family of methods that, given observational data for just two causally related variables, can determine which one is the cause and which one is the effect.
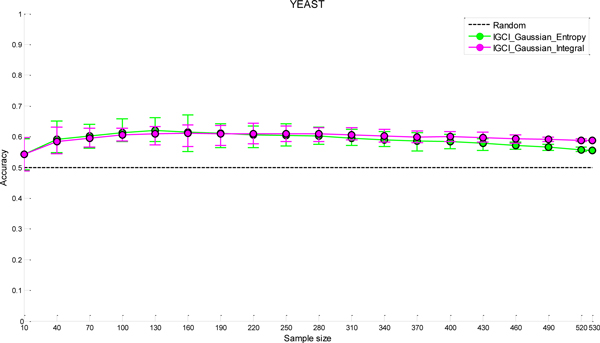
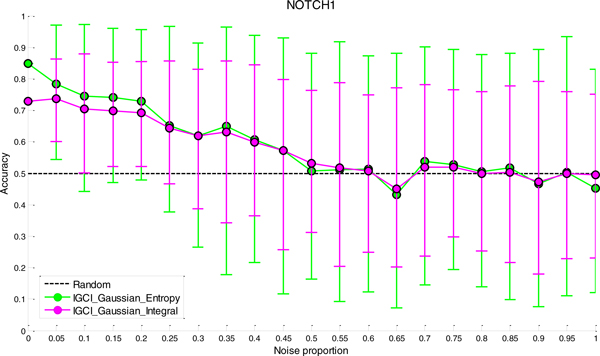
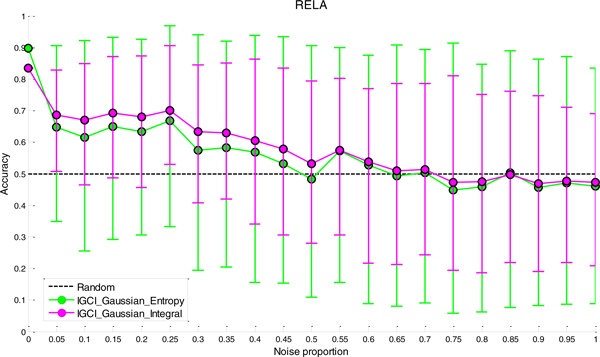
We have found that a particular family of causal orientation methods (IGCI Gaussian) is often able to accurately infer directionality of causal interactions, and that these methods usually outperform other causal orientation techniques. We also introduced a novel ensemble technique for causal orientation that combines decisions of individual causal orientation methods. The ensemble method was found to be more accurate than any best individual causal orientation method in the tested data.
This work represents a first step towards establishing context for practical use of causal orientation methods in the genomics domain. We have found that some causal orientation methodologies yield accurate predictions of causal orientation in genomics data, and we have improved on this capability with a novel ensemble method. Our results suggest that these methods have the potential to facilitate reconstruction of molecular pathways by minimizing the number of required randomized experiments to find causal directionality and by avoiding experiments that are infeasible and/or unethical.
发现分子途径是一个具有挑战性的问题,其解决方案依赖于在基因组学数据中识别因果分子相互作用。可以使用随机实验来发现因果分子相互作用;但是,这样的实验通常成本高昂、不可行或不道德。幸运的是,从观察数据中推断因果相互作用的算法已经开发了几十年,主要在定量科学中,其中许多最近已应用于基因组学数据。虽然这些算法可以推断涉及分子变量之间的无向因果相互作用(即,不指定哪个是原因,哪个是结果),但直到最近,因果定向所有推断出的分子相互作用都被认为是一个无法解决的问题。在这项工作中,我们使用三个不同生物体中的转录因子-靶基因调控相互作用来评估一组新的方法,这些方法给定仅两个因果相关变量的观察数据,就可以确定哪个是原因,哪个是结果。
我们发现,一组特定的因果定向方法(IGCI 高斯)通常能够准确推断因果相互作用的方向,并且这些方法通常优于其他因果定向技术。我们还引入了一种用于因果定向的新集成技术,该技术将单个因果定向方法的决策进行组合。在测试数据中,集合方法比任何最佳的单个因果定向方法都更准确。
这项工作代表了在基因组学领域建立因果定向方法实际应用背景的第一步。我们发现,一些因果定向方法在基因组学数据中能够准确预测因果定向,并且我们通过一种新颖的集成方法提高了这一能力。我们的结果表明,这些方法有可能通过减少找到因果方向所需的随机实验数量,并避免不可行和/或不道德的实验,从而促进分子途径的重建。