Department of Psychology and Neuroscience Institute, Princeton University, Princeton, New Jersey, United States of America.
PLoS One. 2012;7(12):e49949. doi: 10.1371/journal.pone.0049949. Epub 2012 Dec 19.
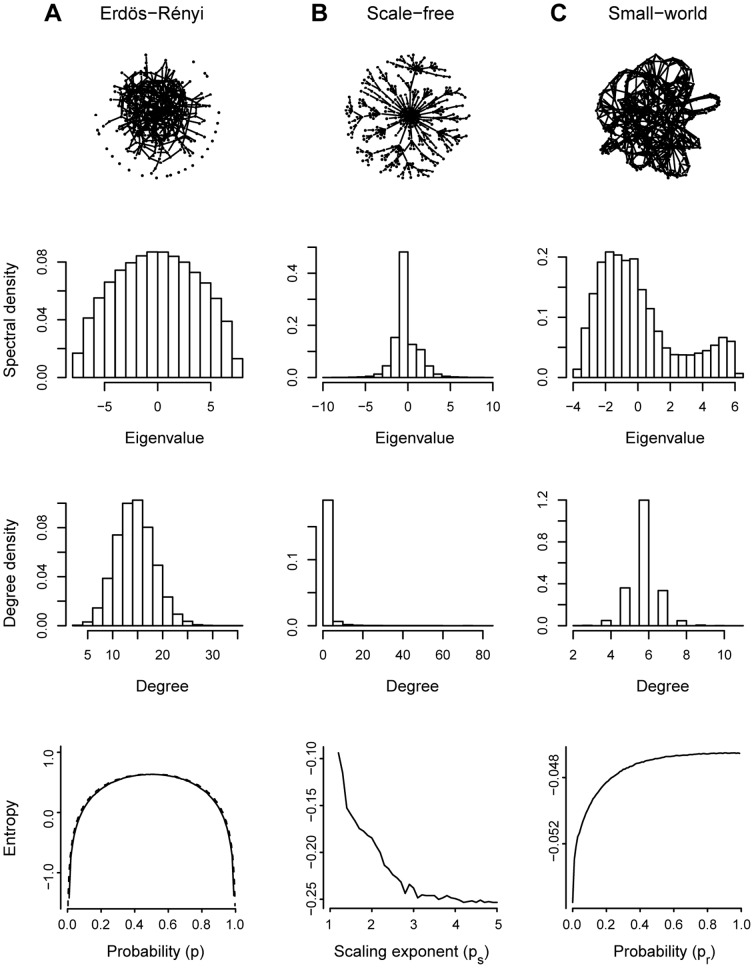
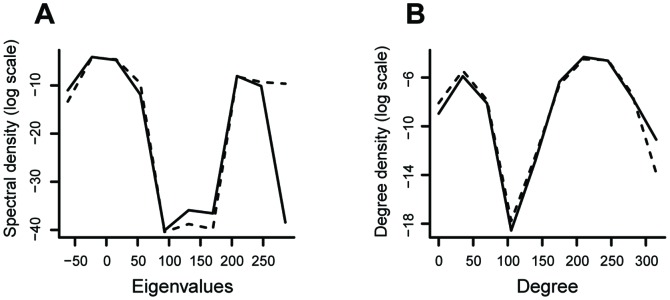
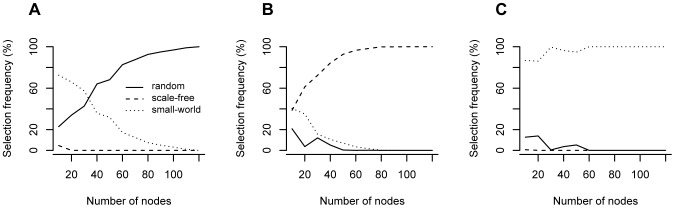
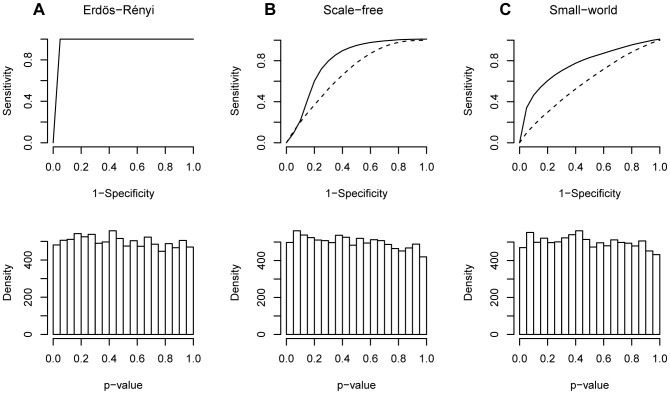
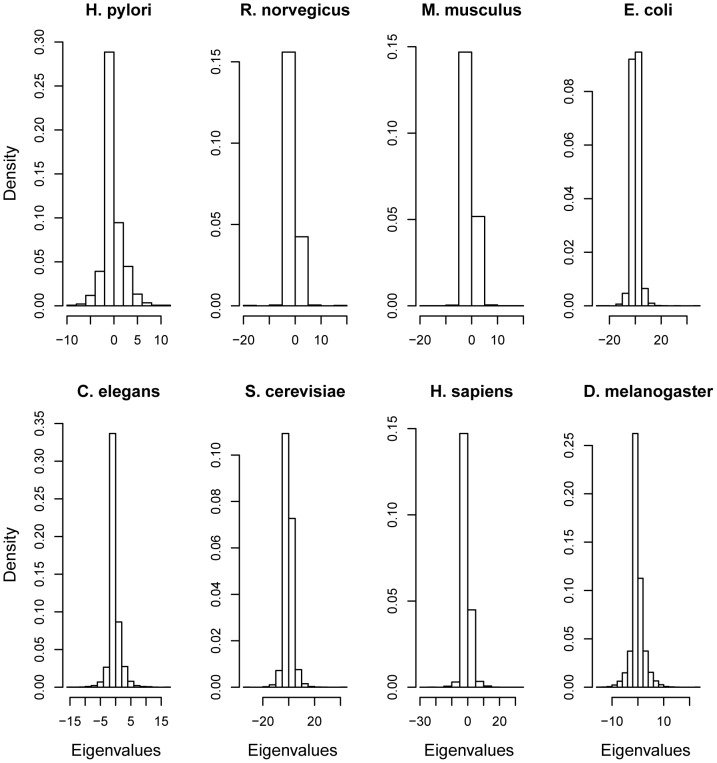
The brain's structural and functional systems, protein-protein interaction, and gene networks are examples of biological systems that share some features of complex networks, such as highly connected nodes, modularity, and small-world topology. Recent studies indicate that some pathologies present topological network alterations relative to norms seen in the general population. Therefore, methods to discriminate the processes that generate the different classes of networks (e.g., normal and disease) might be crucial for the diagnosis, prognosis, and treatment of the disease. It is known that several topological properties of a network (graph) can be described by the distribution of the spectrum of its adjacency matrix. Moreover, large networks generated by the same random process have the same spectrum distribution, allowing us to use it as a "fingerprint". Based on this relationship, we introduce and propose the entropy of a graph spectrum to measure the "uncertainty" of a random graph and the Kullback-Leibler and Jensen-Shannon divergences between graph spectra to compare networks. We also introduce general methods for model selection and network model parameter estimation, as well as a statistical procedure to test the nullity of divergence between two classes of complex networks. Finally, we demonstrate the usefulness of the proposed methods by applying them to (1) protein-protein interaction networks of different species and (2) on networks derived from children diagnosed with Attention Deficit Hyperactivity Disorder (ADHD) and typically developing children. We conclude that scale-free networks best describe all the protein-protein interactions. Also, we show that our proposed measures succeeded in the identification of topological changes in the network while other commonly used measures (number of edges, clustering coefficient, average path length) failed.
大脑的结构和功能系统、蛋白质-蛋白质相互作用以及基因网络是具有复杂网络某些特征的生物系统的示例,例如高度连接的节点、模块性和小世界拓扑结构。最近的研究表明,与一般人群中的正常情况相比,某些病理学存在拓扑网络改变。因此,区分产生不同网络(例如正常和疾病)的过程的方法可能对疾病的诊断、预后和治疗至关重要。已知网络(图)的几个拓扑性质可以通过其邻接矩阵的频谱分布来描述。此外,由相同随机过程生成的大型网络具有相同的频谱分布,这使我们可以将其用作“指纹”。基于这种关系,我们引入并提出了图频谱的熵来衡量随机图的“不确定性”,以及图频谱之间的 Kullback-Leibler 和 Jensen-Shannon 散度来比较网络。我们还介绍了用于模型选择和网络模型参数估计的一般方法,以及用于测试两个复杂网络之间散度的有效性的统计程序。最后,我们通过将所提出的方法应用于(1)不同物种的蛋白质-蛋白质相互作用网络和(2)源自被诊断为注意力缺陷多动障碍(ADHD)和典型发育的儿童的网络,证明了所提出的方法的有用性。我们得出结论,无标度网络最好地描述了所有蛋白质-蛋白质相互作用。此外,我们表明,我们提出的度量标准成功地识别了网络中的拓扑变化,而其他常用的度量标准(边数、聚类系数、平均路径长度)则没有。