Genome Biology Unit, European Molecular Biology Laboratory, Heidelberg, Germany.
Nucleic Acids Res. 2013 Mar 1;41(5):e65. doi: 10.1093/nar/gks1249. Epub 2013 Jan 7.
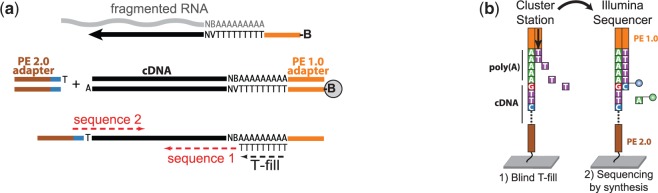
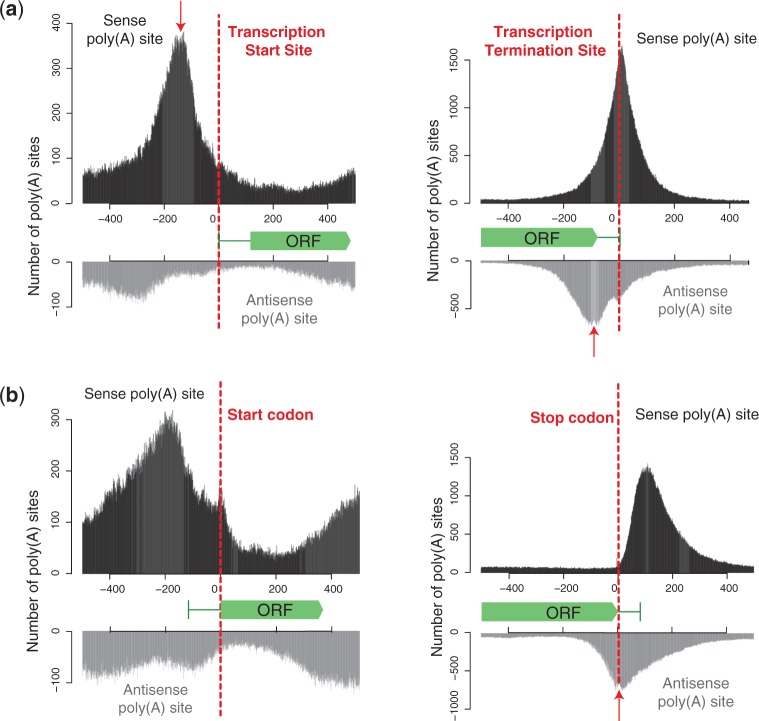
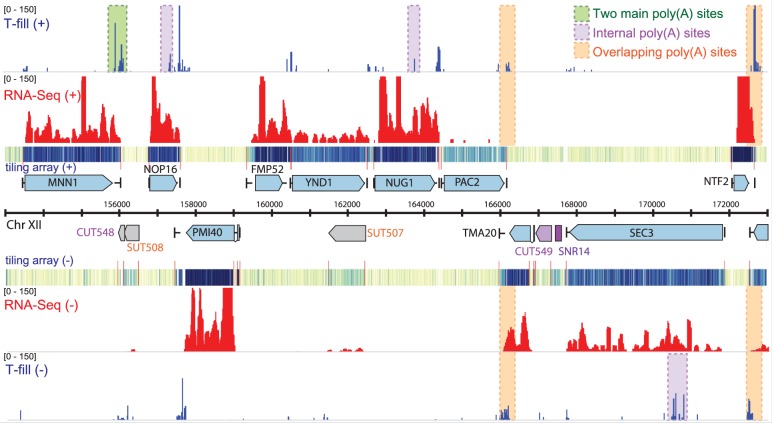
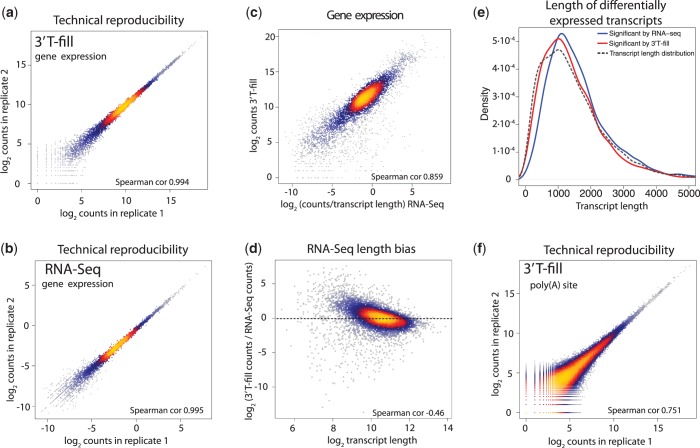
The use of alternative poly(A) sites is common and affects the post-transcriptional fate of mRNA, including its stability, subcellular localization and translation. Here, we present a method to identify poly(A) sites in a genome-wide and strand-specific manner. This method, termed 3'T-fill, initially fills in the poly(A) stretch with unlabeled dTTPs, allowing sequencing to start directly after the poly(A) tail into the 3'-untranslated regions (UTR). Our comparative analysis demonstrates that it outperforms existing protocols in quality and throughput and accurately quantifies RNA levels as only one read is produced from each transcript. We use this method to characterize the diversity of polyadenylation in Saccharomyces cerevisiae, showing that alternative RNA molecules are present even in a genetically identical cell population. Finally, we observe that overlap of convergent 3'-UTRs is frequent but sharply limited by coding regions, suggesting factors that restrict compression of the yeast genome.
非传统 poly(A) 位点的使用很常见,会影响 mRNA 的转录后命运,包括其稳定性、亚细胞定位和翻译。在这里,我们提出了一种在全基因组和链特异性水平上识别 poly(A) 位点的方法。该方法称为 3’T-fill,最初用未标记的 dTTP 填充 poly(A) 延伸,从而可以在 poly(A) 尾巴之后直接进入 3'-非翻译区 (UTR) 进行测序。我们的比较分析表明,它在质量和通量方面优于现有协议,并且由于每个转录物只产生一个读段,因此可以准确地定量 RNA 水平。我们使用这种方法来描述酿酒酵母中 polyadenylation 的多样性,表明即使在遗传上相同的细胞群体中,也存在替代的 RNA 分子。最后,我们观察到,趋同的 3'-UTR 重叠很常见,但受到编码区域的强烈限制,这表明存在限制酵母基因组压缩的因素。