Department of Neurology, Center of Excellence for Novel Approaches to Neurotherapeutics, Mount Sinai School of Medicine, New York, NY, USA.
Neurobiol Aging. 2013 Jun;34(6):1581-8. doi: 10.1016/j.neurobiolaging.2012.12.005. Epub 2013 Jan 9.
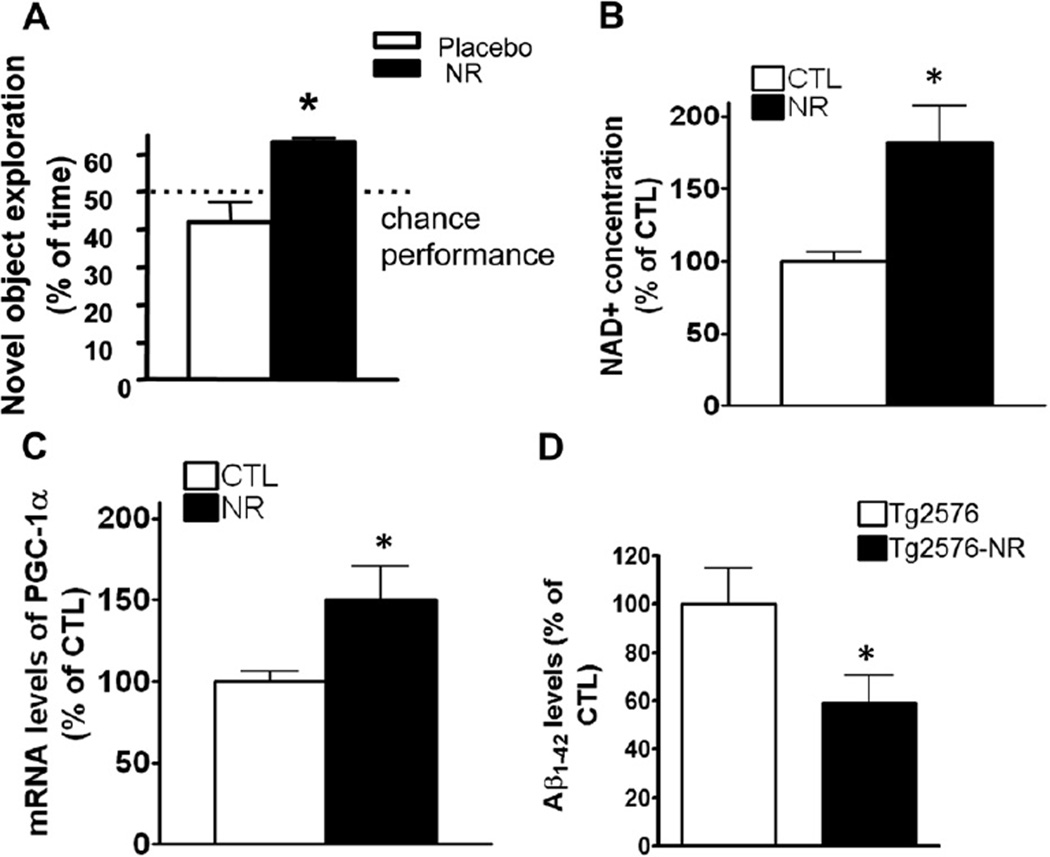
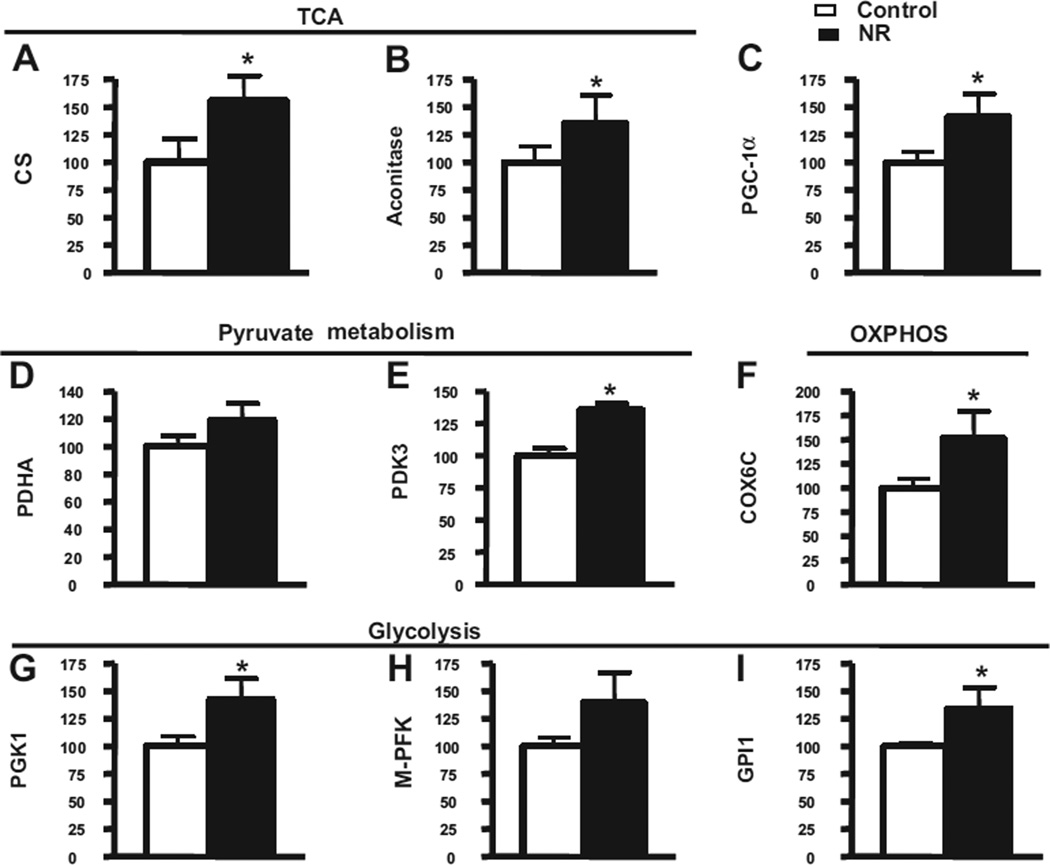
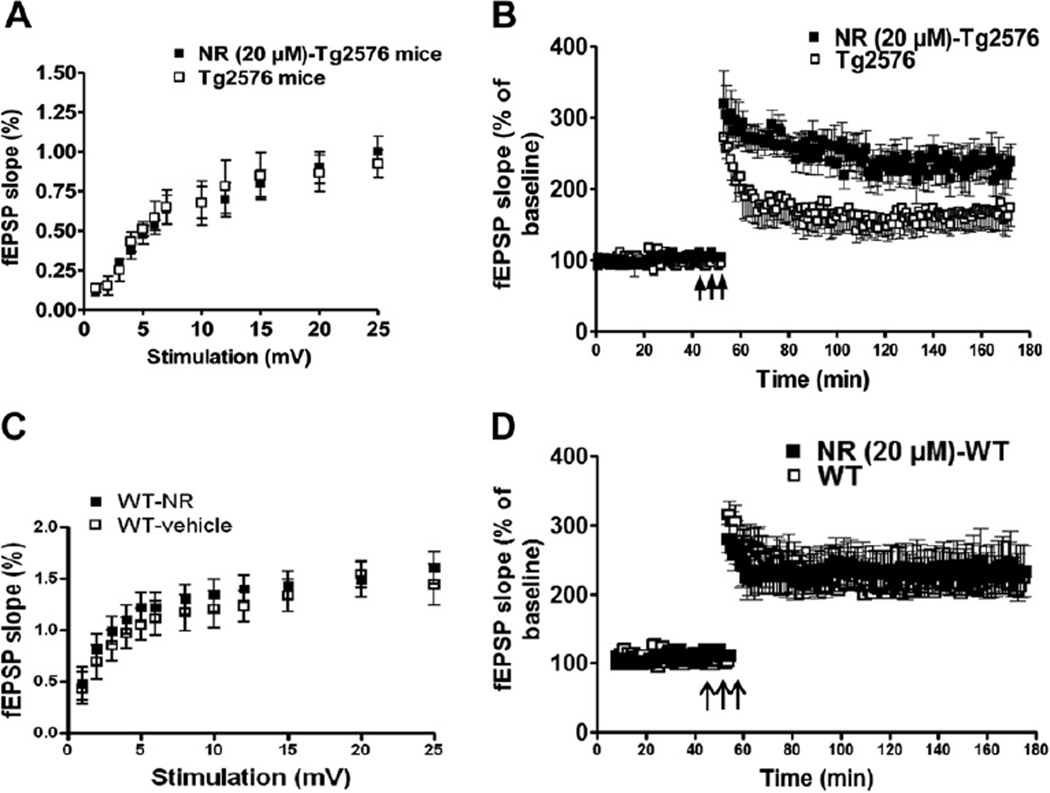
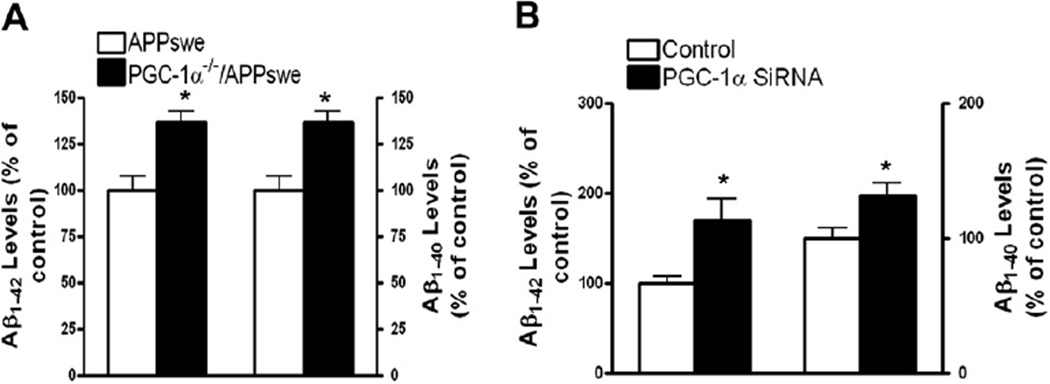
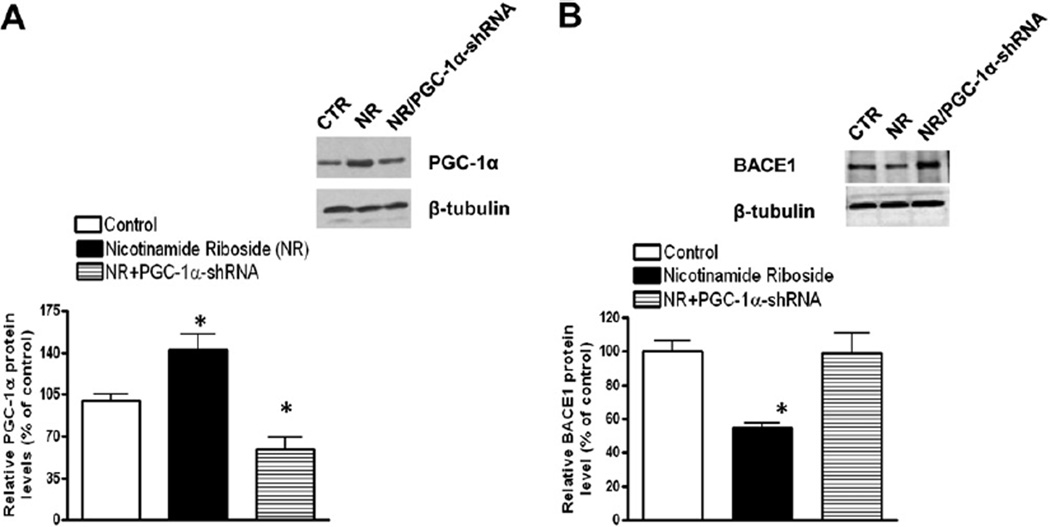
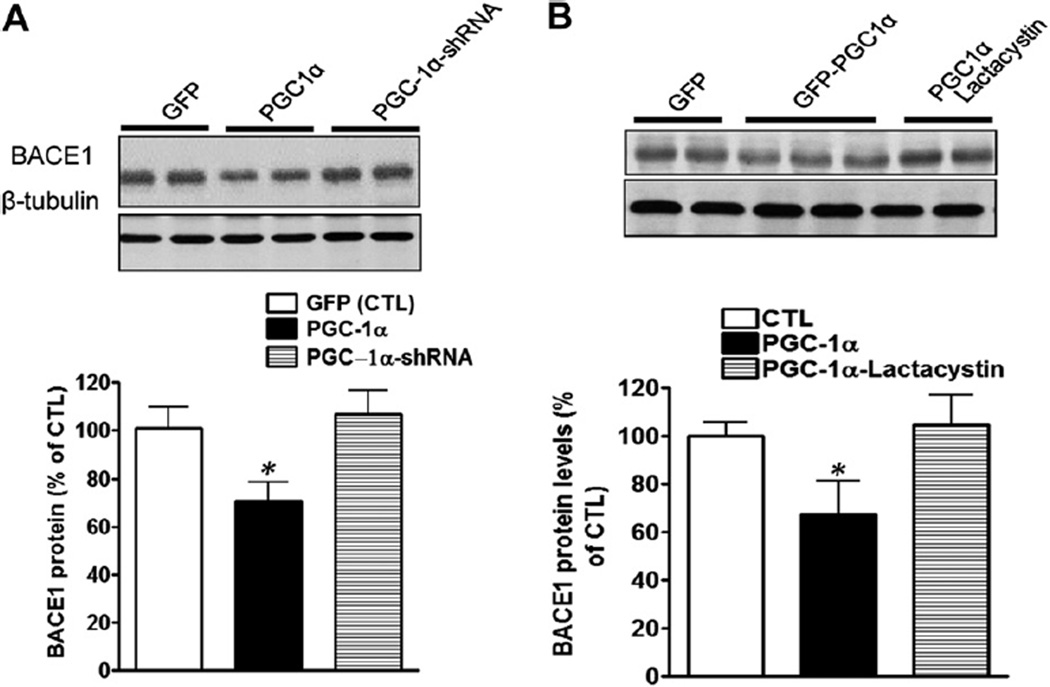
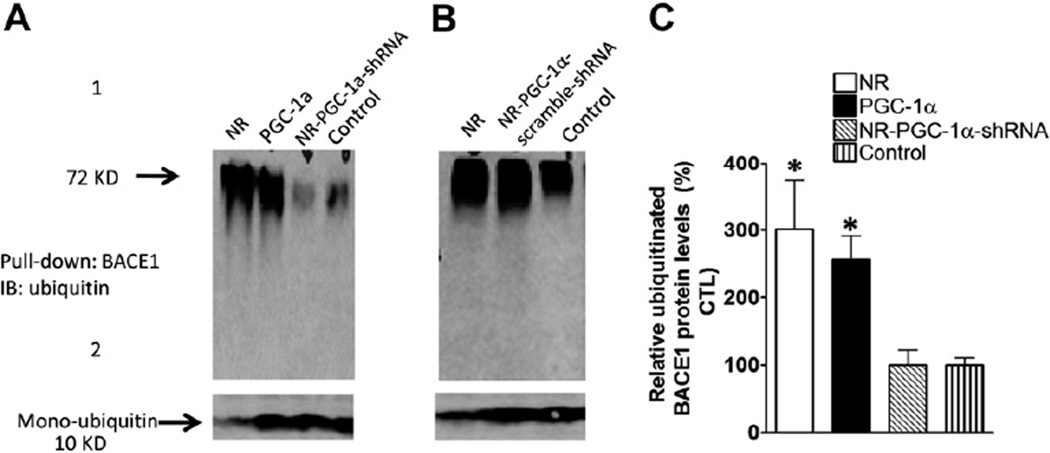
Nicotinamide adenine dinucleotide (NAD)(+), a coenzyme involved in redox activities in the mitochondrial electron transport chain, has been identified as a key regulator of the lifespan-extending effects, and the activation of NAD(+) expression has been linked with a decrease in beta-amyloid (Aβ) toxicity in Alzheimer's disease (AD). Nicotinamide riboside (NR) is a NAD(+) precursor, it promotes peroxisome proliferator-activated receptor-γ coactivator 1 (PGC)-1α expression in the brain. Evidence has shown that PGC-1α is a crucial regulator of Aβ generation because it affects β-secretase (BACE1) degradation. In this study we tested the hypothesis that NR treatment in an AD mouse model could attenuate Aβ toxicity through the activation of PGC-1α-mediated BACE1 degradation. Using the Tg2576 AD mouse model, using in vivo behavioral analyses, biochemistry assays, small hairpin RNA (shRNA) gene silencing and electrophysiological recording, we found (1) dietary treatment of Tg2576 mice with 250 mg/kg/day of NR for 3 months significantly attenuates cognitive deterioration in Tg2576 mice and coincides with an increase in the steady-state levels of NAD(+) in the cerebral cortex; (2) application of NR to hippocampal slices (10 μM) for 4 hours abolishes the deficits in long-term potentiation recorded in the CA1 region of Tg2576 mice; (3) NR treatment promotes PGC-1α expression in the brain coinciding with enhanced degradation of BACE1 and the reduction of Aβ production in Tg2576 mice. Further in vitro studies confirmed that BACE1 protein content is decreased by NR treatment in primary neuronal cultures derived from Tg2576 embryos, in which BACE1 degradation was prevented by PGC-1α-shRNA gene silencing; and (4) NR treatment and PGC-1α overexpression enhance BACE1 ubiquitination and proteasomal degradation. Our studies suggest that dietary treatment with NR might benefit AD cognitive function and synaptic plasticity, in part by promoting PGC-1α-mediated BACE1 ubiquitination and degradation, thus preventing Aβ production in the brain.
烟酰胺腺嘌呤二核苷酸(NAD)(+),一种参与线粒体电子传递链氧化还原活动的辅酶,已被确定为延长寿命效应的关键调节剂,并且 NAD(+)表达的激活与阿尔茨海默病(AD)中β-淀粉样蛋白(Aβ)毒性的降低有关。烟酰胺核苷(NR)是 NAD(+)的前体,它可促进脑内过氧化物酶体增殖物激活受体-γ共激活因子 1(PGC-1α)的表达。有证据表明,PGC-1α 是 Aβ产生的关键调节剂,因为它影响β-分泌酶(BACE1)的降解。在这项研究中,我们测试了这样一种假设,即 NR 治疗 AD 小鼠模型可以通过激活 PGC-1α 介导的 BACE1 降解来减轻 Aβ 毒性。使用 Tg2576 AD 小鼠模型,通过体内行为分析、生化测定、短发夹 RNA(shRNA)基因沉默和电生理记录,我们发现:(1)用 250mg/kg/天的 NR 对 Tg2576 小鼠进行 3 个月的饮食治疗可显著减轻 Tg2576 小鼠的认知恶化,同时大脑皮层中 NAD(+)的稳态水平增加;(2)将 NR 应用于海马切片(10μM)4 小时可消除 Tg2576 小鼠 CA1 区记录的长时程增强缺陷;(3)NR 治疗可促进大脑中 PGC-1α 的表达,同时增强 BACE1 的降解,减少 Tg2576 小鼠中 Aβ的产生。进一步的体外研究证实,NR 处理可降低源自 Tg2576 胚胎的原代神经元培养物中的 BACE1 蛋白含量,其中 BACE1 降解被 PGC-1α-shRNA 基因沉默所阻止;(4)NR 治疗和 PGC-1α 过表达增强 BACE1 泛素化和蛋白酶体降解。我们的研究表明,NR 的饮食治疗可能有益于 AD 的认知功能和突触可塑性,部分原因是通过促进 PGC-1α 介导的 BACE1 泛素化和降解,从而防止大脑中 Aβ的产生。