Department of Biostatistics and Computational Biology, University of Rochester, 601 Elmwood Avenue, Rochester, NY 14642, USA.
BMC Bioinformatics. 2013 Jan 16;14:6. doi: 10.1186/1471-2105-14-6.
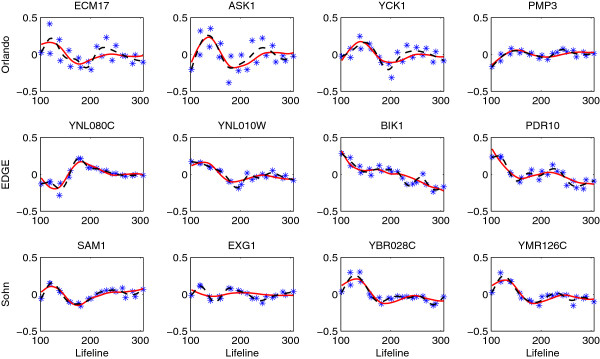
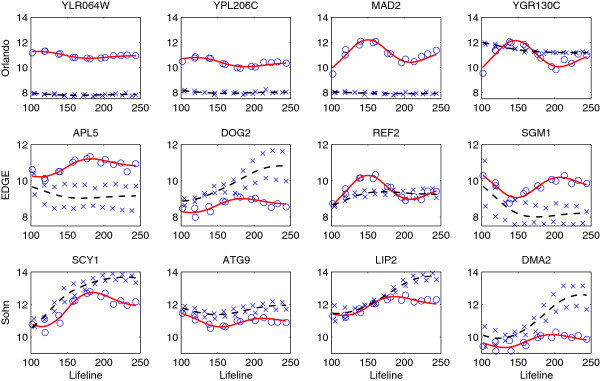
One of the fundamental problems in time course gene expression data analysis is to identify genes associated with a biological process or a particular stimulus of interest, like a treatment or virus infection. Most of the existing methods for this problem are designed for data with longitudinal replicates. But in reality, many time course gene experiments have no replicates or only have a small number of independent replicates.
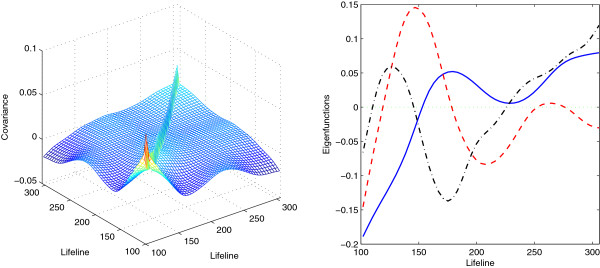
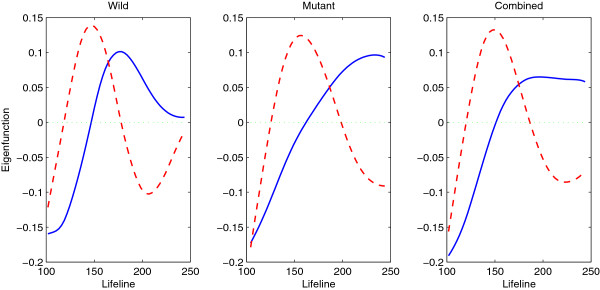
We focus on the case without replicates and propose a new method for identifying differentially expressed genes by incorporating the functional principal component analysis (FPCA) into a hypothesis testing framework. The data-driven eigenfunctions allow a flexible and parsimonious representation of time course gene expression trajectories, leaving more degrees of freedom for the inference compared to that using a prespecified basis. Moreover, the information of all genes is borrowed for individual gene inferences.
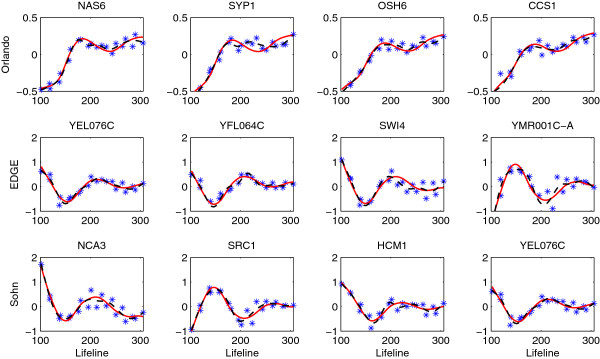
The proposed approach turns out to be more powerful in identifying time course differentially expressed genes compared to the existing methods. The improved performance is demonstrated through simulation studies and a real data application to the Saccharomyces cerevisiae cell cycle data.
在时间序列基因表达数据分析中,一个基本问题是识别与感兴趣的生物过程或特定刺激(如处理或病毒感染)相关的基因。大多数现有的此类问题的方法都是为具有纵向重复的数据集设计的。但实际上,许多时间序列基因实验没有重复,或者只有少数独立的重复。
我们专注于没有重复的情况,并通过将功能主成分分析(FPCA)纳入假设检验框架,提出了一种识别差异表达基因的新方法。数据驱动的特征函数允许灵活且简约地表示时间序列基因表达轨迹,与使用预定义基相比,为推断留出了更多的自由度。此外,还可以为个体基因推断借用所有基因的信息。
与现有方法相比,所提出的方法在识别时间序列差异表达基因方面更有效。通过模拟研究和对酿酒酵母细胞周期数据的实际应用,证明了该方法的性能有所提高。