da Fonseca Paulo G S, Paiva Jorge A P, Almeida Luiz G P, Vasconcelos Ana T R, Freitas Ana T
Instituto de Engenharia de Sistemas e Computadores: Investigação e Desenvolvimento (INESC-ID), R, Alves Redol 9, Lisboa 1000-029, Portugal.
BMC Res Notes. 2013 Jan 22;6:25. doi: 10.1186/1756-0500-6-25.
Sequencing-by-synthesis technologies significantly improve over the Sanger method in terms of speed and cost per base. However, they still usually fail to compete in terms of read length and quality. Current high-throughput implementations of the pyrosequencing technique yield reads whose length approach those of the capillary electrophoresis method. A less obvious question is whether their quality is affected by platform-specific sequencing errors.
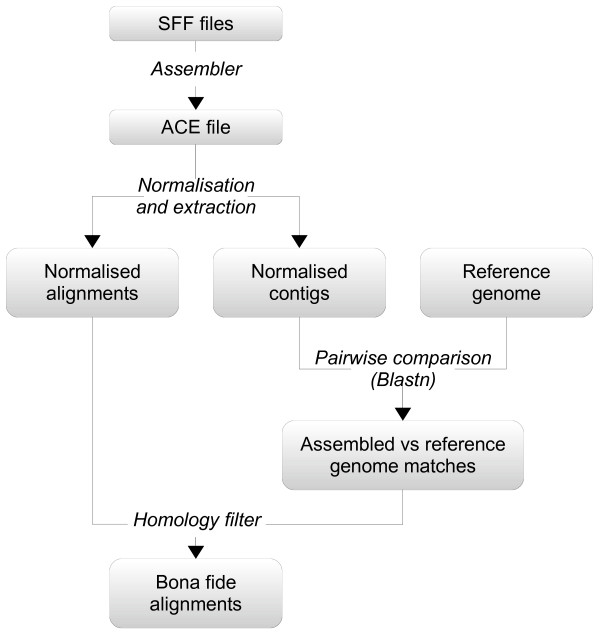
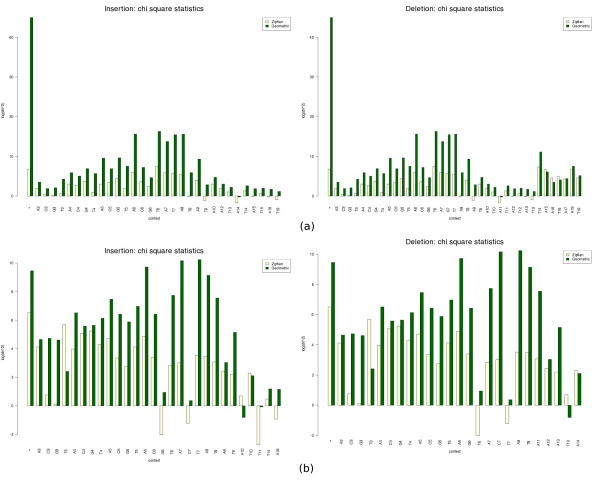
We present an empirical study aimed at assessing the quality and characterising sequencing errors for high throughput pyrosequencing data. We have developed a procedure for extracting sequencing error data from genome assemblies and study their characteristics, in particular the length distribution of indel gaps and their relation to the sequence contexts where they occur. We used this procedure to analyse data from three prokaryotic genomes sequenced with the GS FLX technology. We also compared two models previously employed with success for peptide sequence alignment.
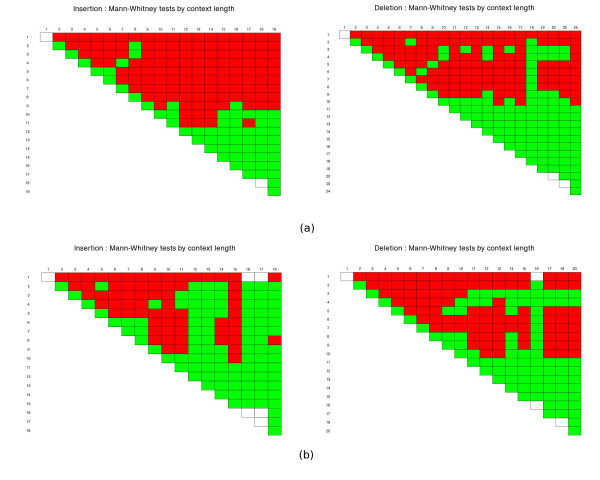
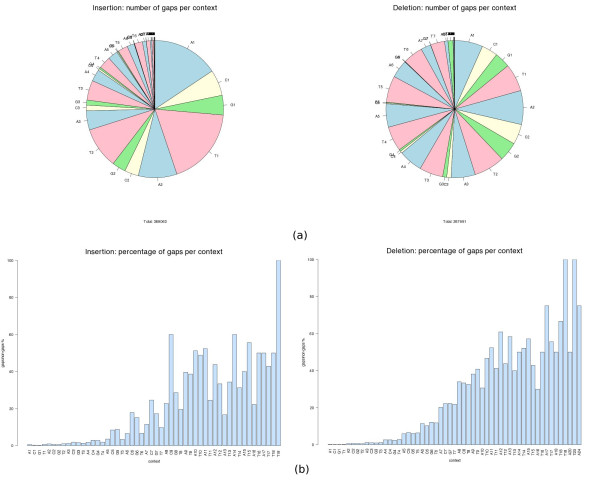
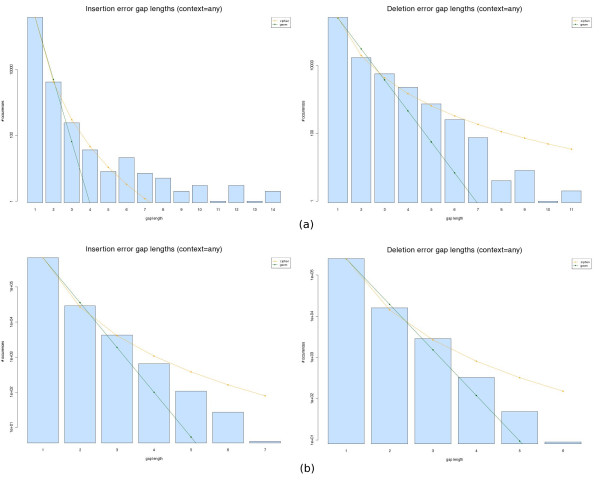
We observed an overall very low error rate in the analysed data, with indel errors being much more abundant than substitutions. We also observed a dependence between the length of the gaps and that of the homopolymer context where they occur. As with protein alignments, a power-law model seems to approximate the indel errors more accurately, although the results are not so conclusive as to justify a depart from the commonly used affine gap penalty scheme. In whichever case, however, our procedure can be used to estimate more realistic error model parameters.
合成测序技术在速度和每碱基成本方面相比桑格法有显著提升。然而,在读取长度和质量方面,它们通常仍无法与之竞争。目前焦磷酸测序技术的高通量实现方式产生的读取长度接近毛细管电泳法。一个不太明显的问题是,其质量是否受到平台特定测序错误的影响。
我们开展了一项实证研究,旨在评估高通量焦磷酸测序数据的质量并表征测序错误。我们开发了一种从基因组组装中提取测序错误数据并研究其特征的程序,特别是插入缺失间隙的长度分布及其与发生位置的序列上下文的关系。我们使用该程序分析了用GS FLX技术测序的三个原核基因组的数据。我们还比较了之前成功用于肽序列比对的两种模型。
我们观察到分析数据中的总体错误率非常低,插入缺失错误比替换错误更为常见。我们还观察到间隙长度与其所在同聚物上下文长度之间的相关性。与蛋白质比对一样,幂律模型似乎能更准确地近似插入缺失错误,尽管结果并不足以确凿到证明要背离常用的仿射间隙罚分方案。然而,无论哪种情况,我们的程序都可用于估计更现实的错误模型参数。