Sanford-Burnham Medical Research Institute, La Jolla, CA 92037, USA.
BMC Genomics. 2013 Feb 12;14:94. doi: 10.1186/1471-2164-14-94.
Genome scale annotation of regulatory interactions and reconstruction of regulatory networks are the crucial problems in bacterial genomics. The Lactobacillales order of bacteria collates various microorganisms having a large economic impact, including both human and animal pathogens and strains used in the food industry. Nonetheless, no systematic genome-wide analysis of transcriptional regulation has been previously made for this taxonomic group.
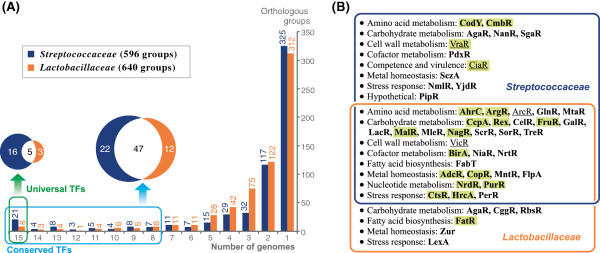
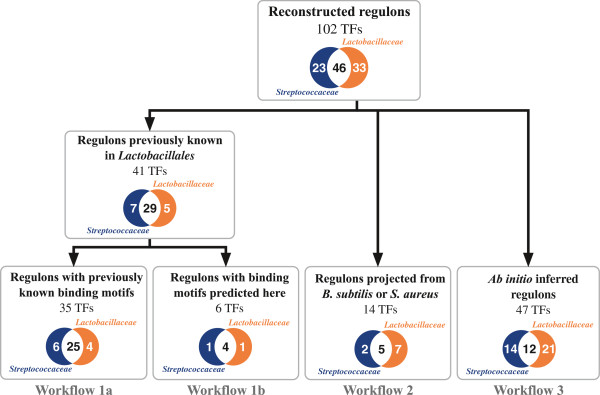
A comparative genomics approach was used for reconstruction of transcriptional regulatory networks in 30 selected genomes of lactic acid bacteria. The inferred networks comprise regulons for 102 orthologous transcription factors (TFs), including 47 novel regulons for previously uncharacterized TFs. Numerous differences between regulatory networks of the Streptococcaceae and Lactobacillaceae groups were described on several levels. The two groups are characterized by substantially different sets of TFs encoded in their genomes. Content of the inferred regulons and structure of their cognate TF binding motifs differ for many orthologous TFs between the two groups. Multiple cases of non-orthologous displacements of TFs that control specific metabolic pathways were reported.
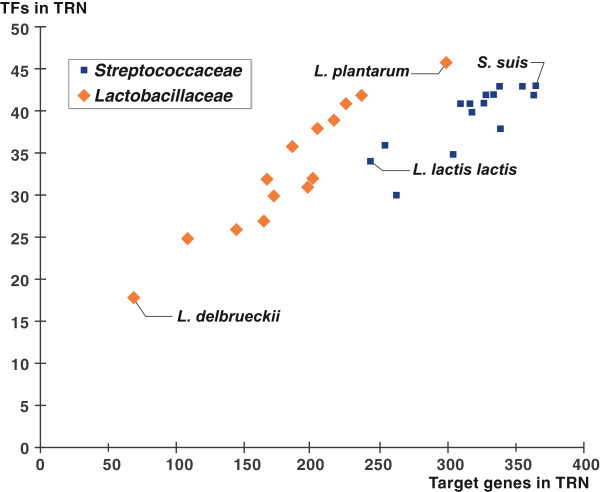
The reconstructed regulatory networks substantially expand the existing knowledge of transcriptional regulation in lactic acid bacteria. In each of 30 studied genomes the obtained regulatory network contains on average 36 TFs and 250 target genes that are mostly involved in carbohydrate metabolism, stress response, metal homeostasis and amino acids biosynthesis. The inferred networks can be used for genetic experiments, functional annotations of genes, metabolic reconstruction and evolutionary analysis. All reconstructed regulons are captured within the Streptococcaceae and Lactobacillaceae collections in the RegPrecise database (http://regprecise.lbl.gov).
调控相互作用的基因组规模注释和调控网络的重建是细菌基因组学中的关键问题。乳杆菌目(Lactobacillales order)汇集了具有巨大经济影响的各种微生物,包括人类和动物病原体以及用于食品工业的菌株。然而,对于这个分类群,以前没有进行过系统的全基因组转录调控分析。
采用比较基因组学方法对 30 株乳酸细菌的选定基因组进行了转录调控网络的重建。推断的网络包含 102 个同源转录因子(TF)的调控子,包括 47 个先前未表征的 TF 的新调控子。在几个层面上描述了链球菌科(Streptococcaceae)和乳杆菌科(Lactobacillaceae)组之间的调控网络的许多差异。这两个组的特点是其基因组中编码的 TF 具有显著不同的集合。推断的调控子的内容及其同源 TF 结合基序的结构在两组之间的许多同源 TF 中存在差异。报告了许多控制特定代谢途径的 TF 非同源置换的情况。
重建的调控网络大大扩展了乳酸细菌中转录调控的现有知识。在所研究的 30 个基因组中的每一个中,获得的调控网络平均包含 36 个 TF 和 250 个靶基因,这些基因主要参与碳水化合物代谢、应激反应、金属稳态和氨基酸生物合成。推断的网络可用于遗传实验、基因的功能注释、代谢重建和进化分析。所有推断的调控子都包含在 RegPrecise 数据库(http://regprecise.lbl.gov)中的链球菌科和乳杆菌科集合中。