Division of Collaboration and Education, Research Center for Zoonosis Control, Hokkaido University, Sapporo-shi, Hokkaido 001-0020, Japan.
DNA Res. 2013 Jun;20(3):209-20. doi: 10.1093/dnares/dst003. Epub 2013 Feb 12.
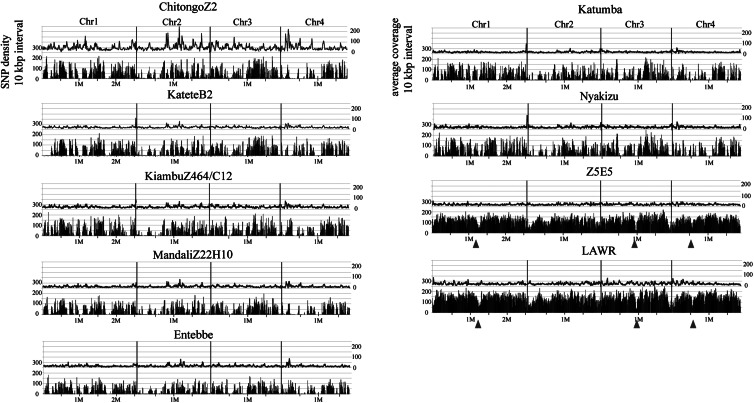
The disease caused by the apicomplexan protozoan parasite Theileria parva, known as East Coast fever or Corridor disease, is one of the most serious cattle diseases in Eastern, Central, and Southern Africa. We performed whole-genome sequencing of nine T. parva strains, including one of the vaccine strains (Kiambu 5), field isolates from Zambia, Uganda, Tanzania, or Rwanda, and two buffalo-derived strains. Comparison with the reference Muguga genome sequence revealed 34 814-121 545 single nucleotide polymorphisms (SNPs) that were more abundant in buffalo-derived strains. High-resolution phylogenetic trees were constructed with selected informative SNPs that allowed the investigation of possible complex recombination events among ancestors of the extant strains. We further analysed the dN/dS ratio (non-synonymous substitutions per non-synonymous site divided by synonymous substitutions per synonymous site) for 4011 coding genes to estimate potential selective pressure. Genes under possible positive selection were identified that may, in turn, assist in the identification of immunogenic proteins or vaccine candidates. This study elucidated the phylogeny of T. parva strains based on genome-wide SNPs analysis with prediction of possible past recombination events, providing insight into the migration, diversification, and evolution of this parasite species in the African continent.
由顶复门原生动物寄生虫泰勒虫引起的疾病,称为东非热或走廊病,是东非、中非和南非最严重的牛病之一。我们对包括一种疫苗株(基安布 5 号)在内的 9 株泰勒虫株进行了全基因组测序,这些株源自赞比亚、乌干达、坦桑尼亚或卢旺达的田间分离株,以及 2 株水牛衍生株。与参考的 Muguga 基因组序列进行比较,发现了 34814-121545 个单核苷酸多态性(SNP),这些 SNP 在水牛衍生株中更为丰富。我们选择了具有信息量的 SNP 构建了高分辨率的系统发育树,这允许对现存株系祖先中可能存在的复杂重组事件进行调查。我们进一步分析了 4011 个编码基因的 dN/dS 比值(非同义替换数除以非同义替换位点数与同义替换数除以同义替换位点数),以估计潜在的选择压力。鉴定出可能受到正选择的基因,这反过来可能有助于鉴定免疫原性蛋白或疫苗候选物。本研究基于全基因组 SNP 分析阐明了泰勒虫株的系统发育,并预测了可能发生的过去重组事件,为了解这种寄生虫在非洲大陆的迁移、多样化和进化提供了线索。