Jia Wenlong, Qiu Kunlong, He Minghui, Song Pengfei, Zhou Quan, Zhou Feng, Yu Yuan, Zhu Dandan, Nickerson Michael L, Wan Shengqing, Liao Xiangke, Zhu Xiaoqian, Peng Shaoliang, Li Yingrui, Wang Jun, Guo Guangwu
Genome Biol. 2013 Feb 14;14(2):R12. doi: 10.1186/gb-2013-14-2-r12.
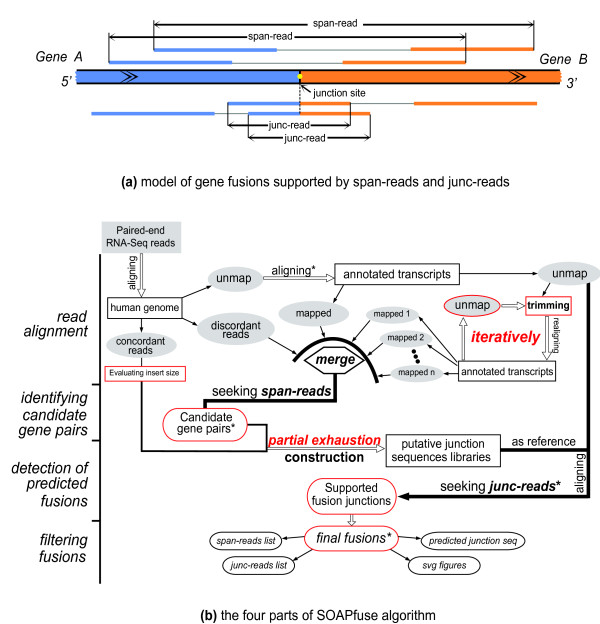
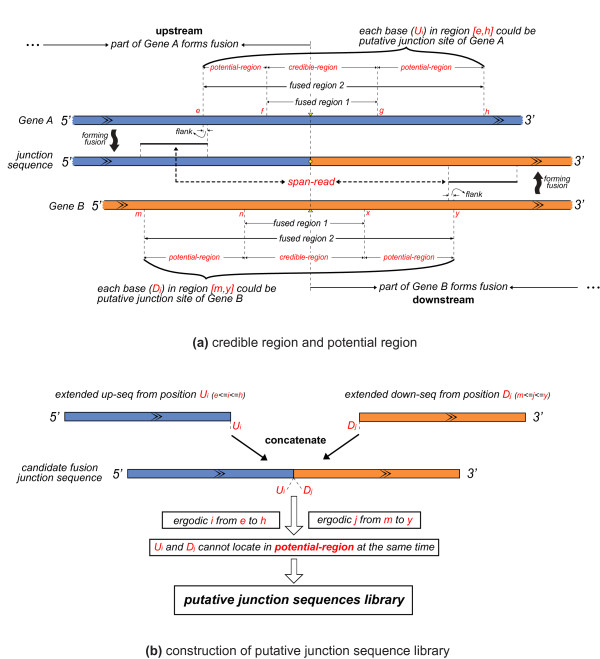
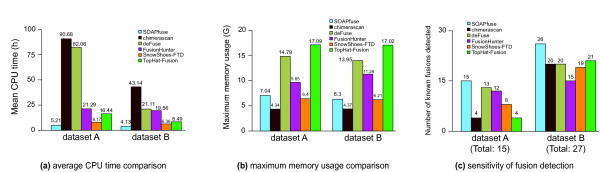
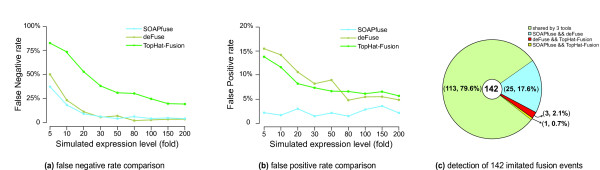
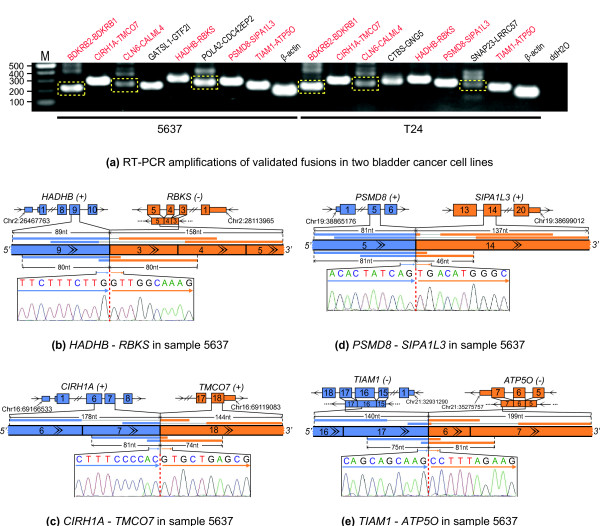
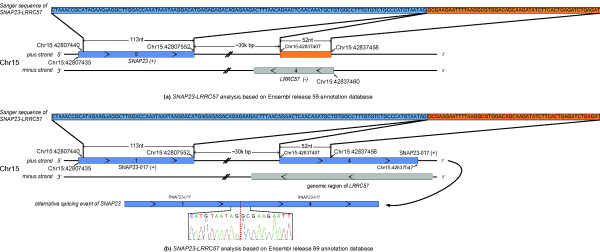
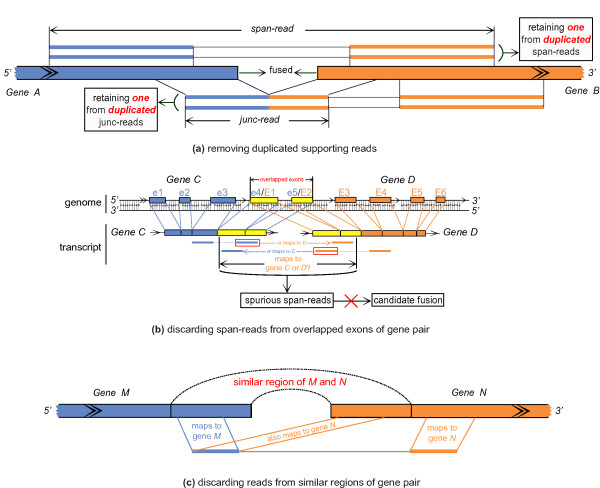
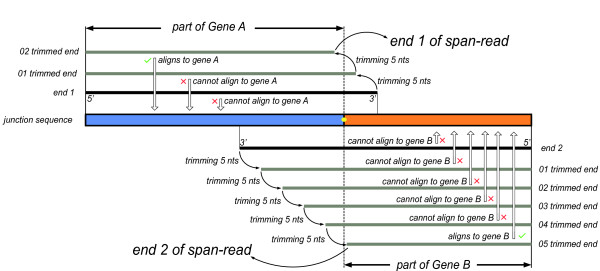
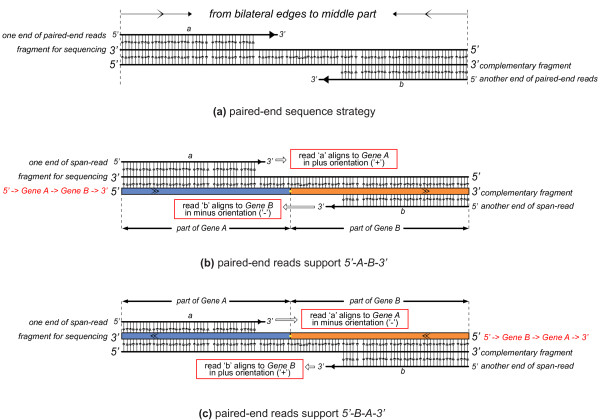
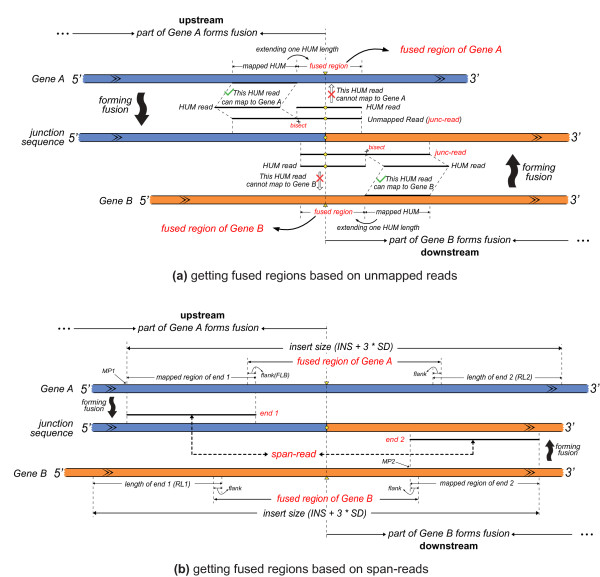
We have developed a new method, SOAPfuse, to identify fusion transcripts from paired-end RNA-Seq data. SOAPfuse applies an improved partial exhaustion algorithm to construct a library of fusion junction sequences, which can be used to efficiently identify fusion events, and employs a series of filters to nominate high-confidence fusion transcripts. Compared with other released tools, SOAPfuse achieves higher detection efficiency and consumed less computing resources. We applied SOAPfuse to RNA-Seq data from two bladder cancer cell lines, and confirmed 15 fusion transcripts, including several novel events common to both cell lines. SOAPfuse is available at http://soap.genomics.org.cn/soapfuse.html.
我们开发了一种新方法SOAPfuse,用于从双末端RNA测序数据中识别融合转录本。SOAPfuse应用一种改进的部分穷举算法来构建融合连接序列文库,该文库可用于高效识别融合事件,并采用一系列筛选器来筛选高可信度的融合转录本。与其他已发布的工具相比,SOAPfuse具有更高的检测效率且消耗更少的计算资源。我们将SOAPfuse应用于来自两种膀胱癌细胞系的RNA测序数据,并确认了15个融合转录本,包括几种两种细胞系共有的新事件。可通过http://soap.genomics.org.cn/soapfuse.html获取SOAPfuse。