Department of Medical Epidemiology and Biostatistics, Karolinska Institutet, Nobels väg 12A, Stockholm, 17177, Sweden.
Department of Computational Sciences and Engineering, VNU University of Engineering and Technology, Xuan Thuy, 144, Hanoi, 84024, Vietnam.
BMC Genomics. 2018 Nov 1;19(1):786. doi: 10.1186/s12864-018-5156-1.
Fusion genes are known to be drivers of many common cancers, so they are potential markers for diagnosis, prognosis or therapy response. The advent of paired-end RNA sequencing enhances our ability to discover fusion genes. While there are available methods, routine analyses of large number of samples are still limited due to high computational demands.
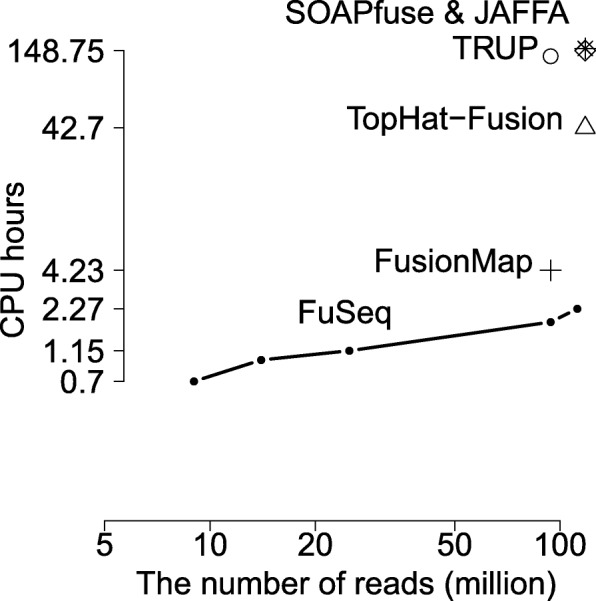
We develop FuSeq, a fast and accurate method to discover fusion genes based on quasi-mapping to quickly map the reads, extract initial candidates from split reads and fusion equivalence classes of mapped reads, and finally apply multiple filters and statistical tests to get the final candidates. We apply FuSeq to four validated datasets: breast cancer, melanoma and glioma datasets, and one spike-in dataset. The results reveal high sensitivity and specificity in all datasets, and compare well against other methods such as FusionMap, TRUP, TopHat-Fusion, SOAPfuse and JAFFA. In terms of computational time, FuSeq is two-fold faster than FusionMap and orders of magnitude faster than the other methods.
With this advantage of less computational demands, FuSeq makes it practical to investigate fusion genes in large numbers of samples. FuSeq is implemented in C++ and R, and available at https://github.com/nghiavtr/FuSeq for non-commercial uses.
融合基因已知是许多常见癌症的驱动因素,因此它们是潜在的诊断、预后或治疗反应的标志物。端对端 RNA 测序的出现提高了我们发现融合基因的能力。虽然有可用的方法,但由于计算需求高,对大量样本进行常规分析仍然受到限制。
我们开发了 FuSeq,这是一种快速准确的方法,基于准映射来发现融合基因,快速映射读取,从分割读取和映射读取的融合等价类中提取初始候选物,最后应用多个过滤器和统计测试来获得最终候选物。我们将 FuSeq 应用于四个经过验证的数据集:乳腺癌、黑色素瘤和神经胶质瘤数据集,以及一个 Spike-in 数据集。结果表明,在所有数据集上均具有较高的灵敏度和特异性,与 FusionMap、TRUP、TopHat-Fusion、SOAPfuse 和 JAFFA 等其他方法相比表现良好。在计算时间方面,FuSeq 比 FusionMap 快两倍,比其他方法快几个数量级。
FuSeq 具有计算需求低的优势,使得在大量样本中研究融合基因成为可能。FuSeq 是用 C++和 R 实现的,可在 https://github.com/nghiavtr/FuSeq 上免费获取,供非商业用途使用。