Phytochemistry and Organic Synthesis Laboratory, School of Pharmacy, Federal University of Rio Grande do Sul, Porto Alegre, 90610-000, Brazil.
Malar J. 2013 Mar 9;12:89. doi: 10.1186/1475-2875-12-89.
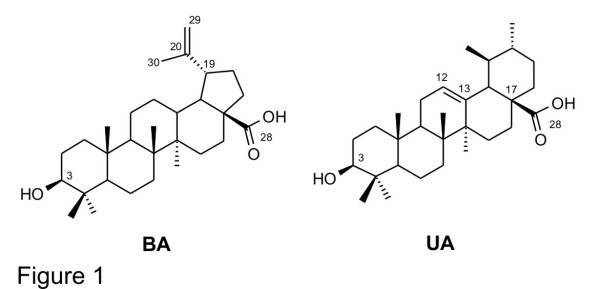
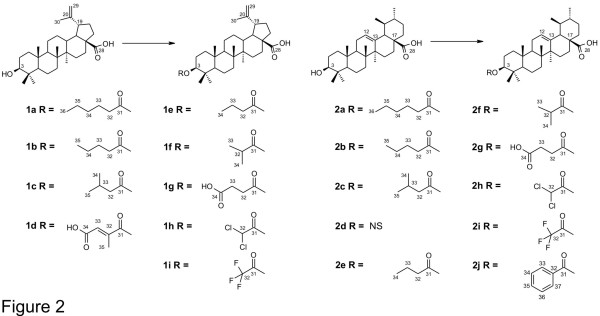
The discovery and development of anti-malarial compounds of plant origin and semisynthetic derivatives thereof, such as quinine (QN) and chloroquine (CQ), has highlighted the importance of these compounds in the treatment of malaria. Ursolic acid analogues bearing an acetyl group at C-3 have demonstrated significant anti-malarial activity. With this in mind, two new series of betulinic acid (BA) and ursolic acid (UA) derivatives with ester groups at C-3 were synthesized in an attempt to improve anti-malarial activity, reduce cytotoxicity, and search for new targets. In vitro activity against CQ-sensitive Plasmodium falciparum 3D7 and an evaluation of cytotoxicity in a mammalian cell line (HEK293T) are reported. Furthermore, two possible mechanisms of action of anti-malarial compounds have been evaluated: effects on mitochondrial membrane potential (ΔΨm) and inhibition of β-haematin formation.
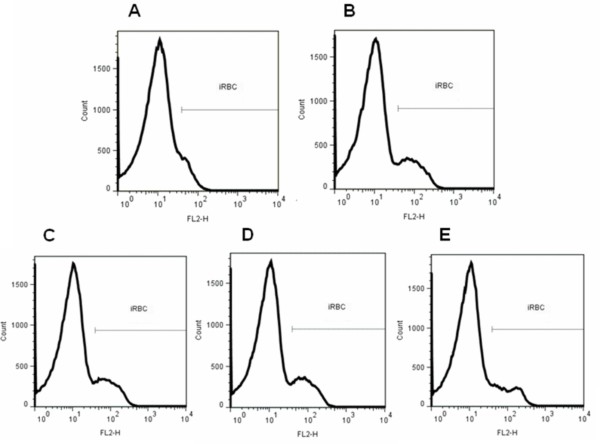
Among the 18 derivatives synthesized, those having shorter side chains were most effective against CQ-sensitive P. falciparum 3D7, and were non-cytotoxic. These derivatives were three to five times more active than BA and UA. A DiOC(6)(3) ΔΨm assay showed that mitochondria are not involved in their mechanism of action. Inhibition of β-haematin formation by the active derivatives was weaker than with CQ. Compounds of the BA series were generally more active against P. falciparum 3D7 than those of the UA series.
Three new anti-malarial prototypes were obtained from natural sources through an easy and relatively inexpensive synthesis. They represent an alternative for new lead compounds for anti-malarial chemotherapy.
从植物来源的抗疟化合物和半合成衍生物,如奎宁(QN)和氯喹(CQ)的发现和开发,凸显了这些化合物在治疗疟疾中的重要性。在 C-3 位带有乙酰基的熊果酸类似物表现出显著的抗疟活性。考虑到这一点,我们合成了两个新系列的桦木酸(BA)和熊果酸(UA)衍生物,它们在 C-3 位带有酯基,旨在提高抗疟活性、降低细胞毒性,并寻找新的靶点。报告了对 CQ 敏感的恶性疟原虫 3D7 的体外活性,并评估了在哺乳动物细胞系(HEK293T)中的细胞毒性。此外,还评估了两种抗疟化合物的可能作用机制:对线粒体膜电位(ΔΨm)的影响和对β-血红素形成的抑制作用。
在所合成的 18 种衍生物中,那些侧链较短的衍生物对 CQ 敏感的恶性疟原虫 3D7 最为有效,且无细胞毒性。这些衍生物比 BA 和 UA 活性高三到五倍。DiOC(6)(3)ΔΨm 测定表明线粒体不参与其作用机制。活性衍生物对β-血红素形成的抑制作用弱于 CQ。BA 系列的化合物通常比 UA 系列对恶性疟原虫 3D7 的活性更高。
通过简单且相对廉价的合成,从天然来源获得了三种新的抗疟原型。它们代表了抗疟化疗新先导化合物的替代物。