Hubrecht Institute/KNAW and University Medical Center Utrecht, Uppsalalaan 8, Utrecht 3584 CT, The Netherlands.
BMC Genomics. 2013 Apr 16;14:257. doi: 10.1186/1471-2164-14-257.
Paired-tag sequencing approaches are commonly used for the analysis of genome structure. However, mammalian genomes have a complex organization with a variety of repetitive elements that complicate comprehensive genome-wide analyses.
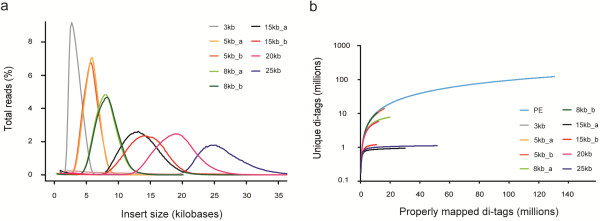
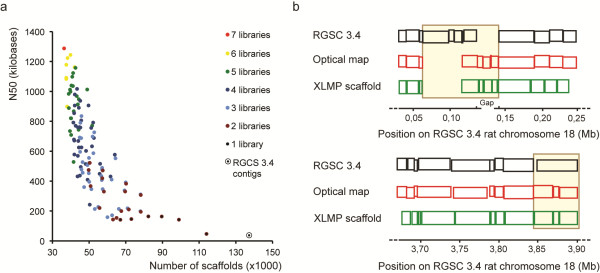
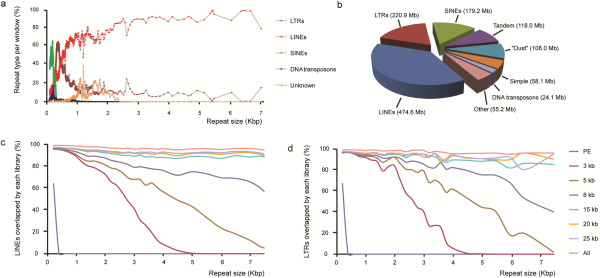
Here, we systematically assessed the utility of paired-end and mate-pair (MP) next-generation sequencing libraries with insert sizes ranging from 170 bp to 25 kb, for genome coverage and for improving scaffolding of a mammalian genome (Rattus norvegicus). Despite a lower library complexity, large insert MP libraries (20 or 25 kb) provided very high physical genome coverage and were found to efficiently span repeat elements in the genome. Medium-sized (5, 8 or 15 kb) MP libraries were much more efficient for genome structure analysis than the more commonly used shorter insert paired-end and 3 kb MP libraries. Furthermore, the combination of medium- and large insert libraries resulted in a 3-fold increase in N50 in scaffolding processes. Finally, we show that our data can be used to evaluate and improve contig order and orientation in the current rat reference genome assembly.
We conclude that applying combinations of mate-pair libraries with insert sizes that match the distributions of repetitive elements improves contig scaffolding and can contribute to the finishing of draft genomes.
配对标签测序方法常用于基因组结构分析。然而,哺乳动物基因组具有复杂的组织,存在多种重复元件,这使得全面的全基因组分析变得复杂。
在这里,我们系统地评估了插入大小范围为 170bp 至 25kb 的 2 种下一代测序文库(即:配对末端文库和 mate-pair 文库)在基因组覆盖度和提高哺乳动物基因组(即:挪威鼠)组装支架方面的应用。尽管文库复杂度较低,但较大的插入 MP 文库(20kb 或 25kb)提供了非常高的物理基因组覆盖率,并且被发现能够有效地跨越基因组中的重复元件。与更常用的较短插入的配对末端文库和 3kb 的 MP 文库相比,中等大小(5kb、8kb 或 15kb)的 MP 文库更有利于基因组结构分析。此外,中等和较大插入文库的组合导致支架过程中 N50 增加了 3 倍。最后,我们表明,我们的数据可用于评估和改善当前大鼠参考基因组组装中连续序列的顺序和方向。
我们的结论是,应用与重复元件分布相匹配的 mate-pair 文库组合可提高连续序列的支架质量,并有助于完成基因组草图。