Department of Biostatistics and Programming, Mail Stop 55C-305A, 55 Corporate Drive, Sanofi, Bridgewater, NJ 08807, USA.
BMC Bioinformatics. 2013 Apr 19;14:132. doi: 10.1186/1471-2105-14-132.
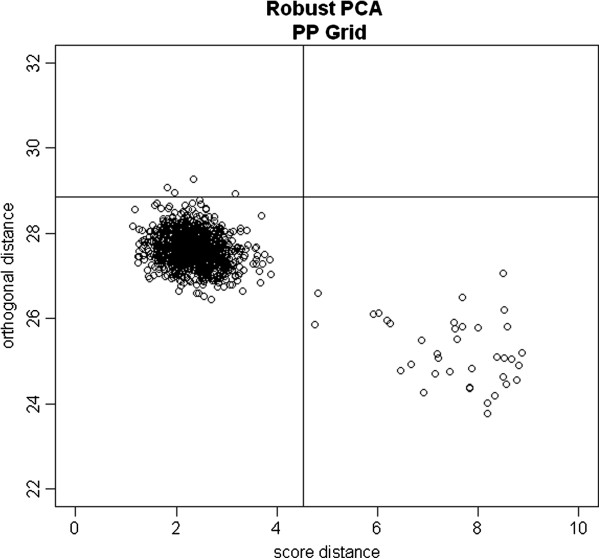
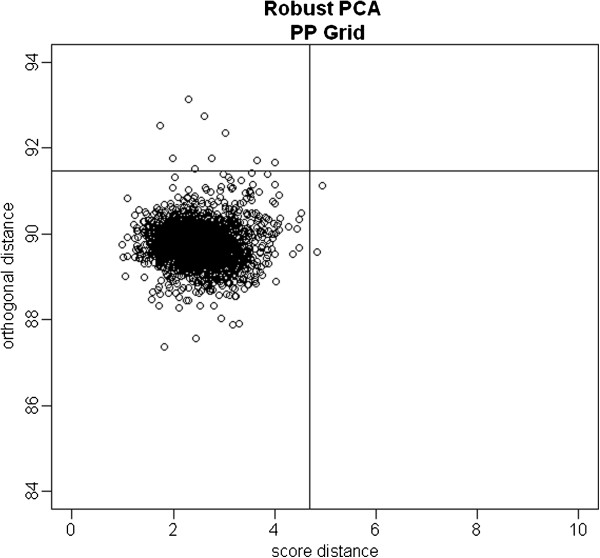
Genome-wide association studies can provide novel insights into diseases of interest, as well as to the responsiveness of an individual to specific treatments. In such studies, it is very important to correct for population stratification, which refers to allele frequency differences between cases and controls due to systematic ancestry differences. Population stratification can cause spurious associations if not adjusted properly. The principal component analysis (PCA) method has been relied upon as a highly useful methodology to adjust for population stratification in these types of large-scale studies. Recently, the linear mixed model (LMM) has also been proposed to account for family structure or cryptic relatedness. However, neither of these approaches may be optimal in properly correcting for sample structures in the presence of subject outliers.
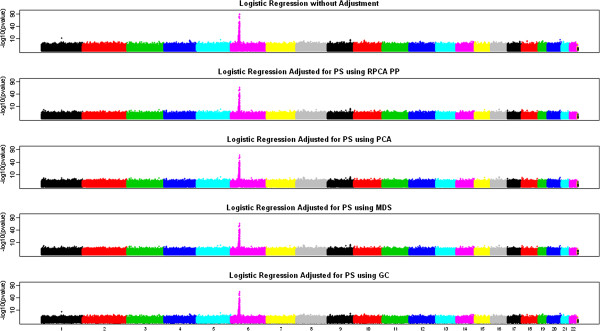
We propose to use robust PCA combined with k-medoids clustering to deal with population stratification. This approach can adjust for population stratification for both continuous and discrete populations with subject outliers, and it can be considered as an extension of the PCA method and the multidimensional scaling (MDS) method. Through simulation studies, we compare the performance of our proposed methods with several widely used stratification methods, including PCA and MDS. We show that subject outliers can greatly influence the analysis results from several existing methods, while our proposed robust population stratification methods perform very well for both discrete and admixed populations with subject outliers. We illustrate the new method using data from a rheumatoid arthritis study.
We demonstrate that subject outliers can greatly influence the analysis result in GWA studies, and propose robust methods for dealing with population stratification that outperform existing population stratification methods in the presence of subject outliers.
全基因组关联研究可以为感兴趣的疾病以及个体对特定治疗的反应提供新的见解。在这类研究中,校正群体分层非常重要,群体分层是指由于系统的祖先差异,病例和对照之间等位基因频率的差异。如果不进行适当调整,群体分层可能会导致虚假关联。主成分分析(PCA)方法已被广泛用作调整这些大规模研究中群体分层的高度有用的方法。最近,线性混合模型(LMM)也被提出用于解释家族结构或隐藏的相关性。然而,在存在个体离群值的情况下,这些方法都可能无法很好地校正样本结构。
我们建议使用稳健 PCA 与 k-中心点聚类相结合来处理群体分层。这种方法可以调整存在个体离群值的连续和离散人群的群体分层,它可以被视为 PCA 方法和多维尺度(MDS)方法的扩展。通过模拟研究,我们将我们提出的方法与几种广泛使用的分层方法(包括 PCA 和 MDS)的性能进行了比较。我们表明,个体离群值会极大地影响几种现有方法的分析结果,而我们提出的稳健的群体分层方法在存在个体离群值的离散和混合人群中表现非常出色。我们使用类风湿关节炎研究的数据说明了新方法。
我们证明了个体离群值会极大地影响 GWA 研究中的分析结果,并提出了稳健的方法来处理群体分层,这些方法在存在个体离群值的情况下优于现有的群体分层方法。