Conomos Matthew P, Miller Michael B, Thornton Timothy A
Department of Biostatistics, University of Washington, Seattle, Washington, 98195, United States of America.
Genet Epidemiol. 2015 May;39(4):276-93. doi: 10.1002/gepi.21896. Epub 2015 Mar 23.
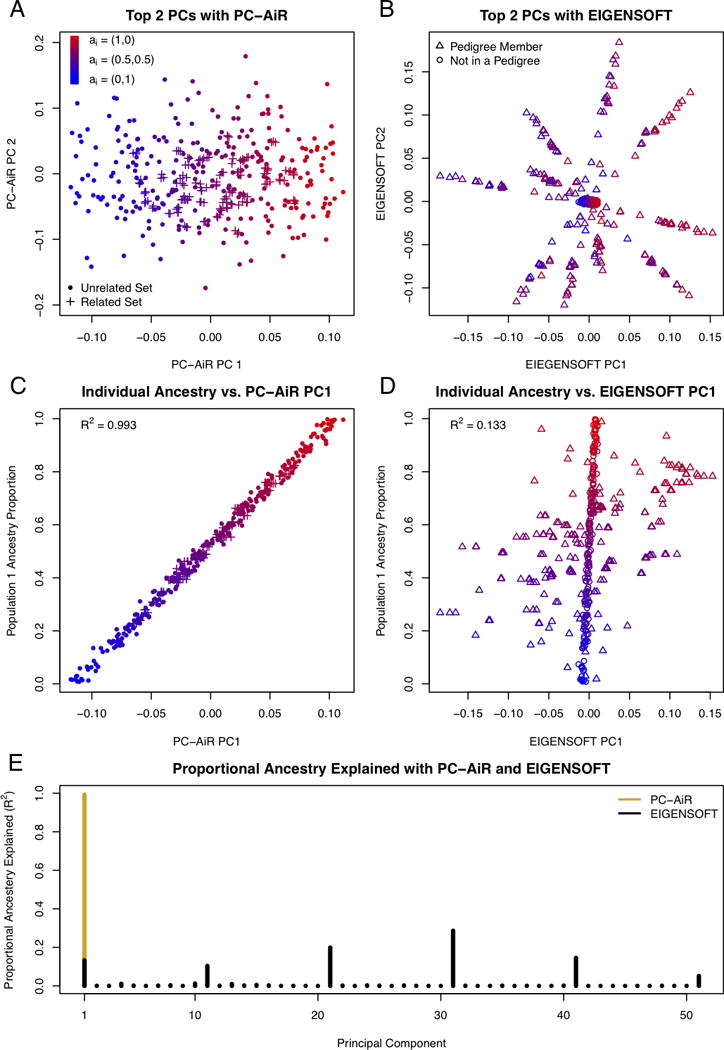
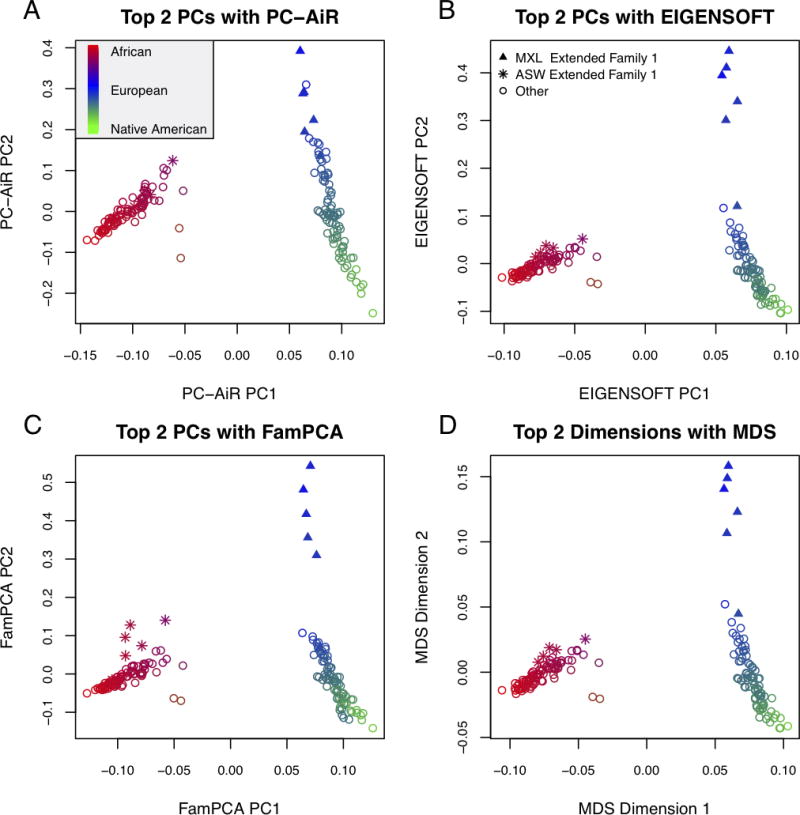
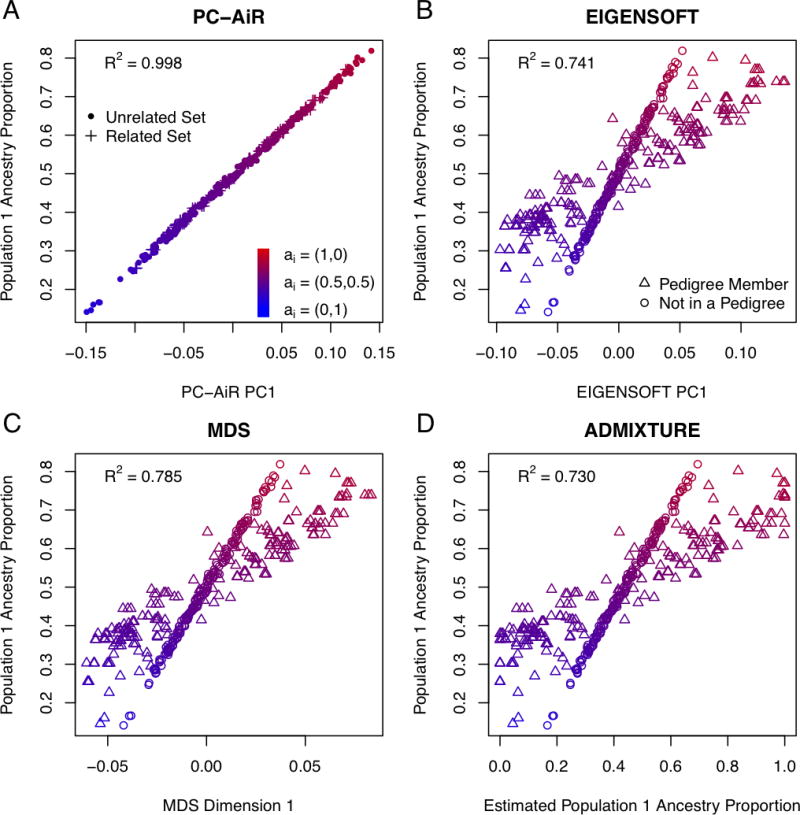
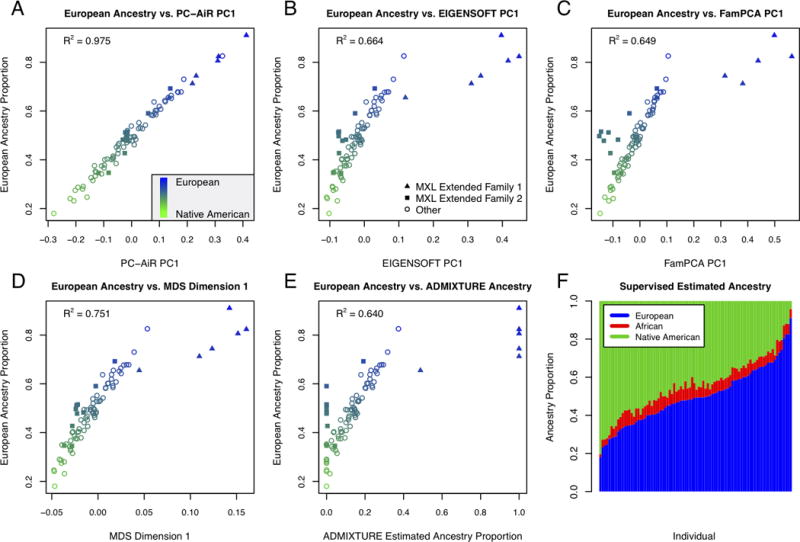
Population structure inference with genetic data has been motivated by a variety of applications in population genetics and genetic association studies. Several approaches have been proposed for the identification of genetic ancestry differences in samples where study participants are assumed to be unrelated, including principal components analysis (PCA), multidimensional scaling (MDS), and model-based methods for proportional ancestry estimation. Many genetic studies, however, include individuals with some degree of relatedness, and existing methods for inferring genetic ancestry fail in related samples. We present a method, PC-AiR, for robust population structure inference in the presence of known or cryptic relatedness. PC-AiR utilizes genome-screen data and an efficient algorithm to identify a diverse subset of unrelated individuals that is representative of all ancestries in the sample. The PC-AiR method directly performs PCA on the identified ancestry representative subset and then predicts components of variation for all remaining individuals based on genetic similarities. In simulation studies and in applications to real data from Phase III of the HapMap Project, we demonstrate that PC-AiR provides a substantial improvement over existing approaches for population structure inference in related samples. We also demonstrate significant efficiency gains, where a single axis of variation from PC-AiR provides better prediction of ancestry in a variety of structure settings than using 10 (or more) components of variation from widely used PCA and MDS approaches. Finally, we illustrate that PC-AiR can provide improved population stratification correction over existing methods in genetic association studies with population structure and relatedness.
利用遗传数据进行群体结构推断,是受群体遗传学和遗传关联研究中的各种应用所推动。对于假定研究参与者无亲缘关系的样本,已经提出了几种方法来识别遗传血统差异,包括主成分分析(PCA)、多维尺度分析(MDS)以及用于比例血统估计的基于模型的方法。然而,许多遗传研究包含有一定亲缘关系的个体,而现有的推断遗传血统的方法在相关样本中会失效。我们提出了一种方法,即PC - AiR,用于在存在已知或潜在亲缘关系的情况下进行稳健的群体结构推断。PC - AiR利用基因组筛选数据和一种高效算法,来识别一个无亲缘关系个体的多样化子集,该子集代表了样本中的所有血统。PC - AiR方法直接对所识别的血统代表性子集进行主成分分析,然后基于遗传相似性预测所有其余个体的变异成分。在模拟研究以及对国际人类基因组单体型图计划(HapMap)项目第三阶段真实数据的应用中,我们证明PC - AiR相对于现有方法,在相关样本的群体结构推断方面有显著改进。我们还证明了显著的效率提升,即在各种结构设置下,PC - AiR的单个变异轴比使用广泛使用的PCA和MDS方法的10个(或更多)变异成分,能更好地预测血统。最后,我们说明在存在群体结构和亲缘关系的遗传关联研究中,PC - AiR相对于现有方法可以提供更好的群体分层校正。