Department of Plant Systems Biology, VIB, Gent, Belgium.
PLoS One. 2013 Apr 17;8(4):e55814. doi: 10.1371/journal.pone.0055814. Print 2013.
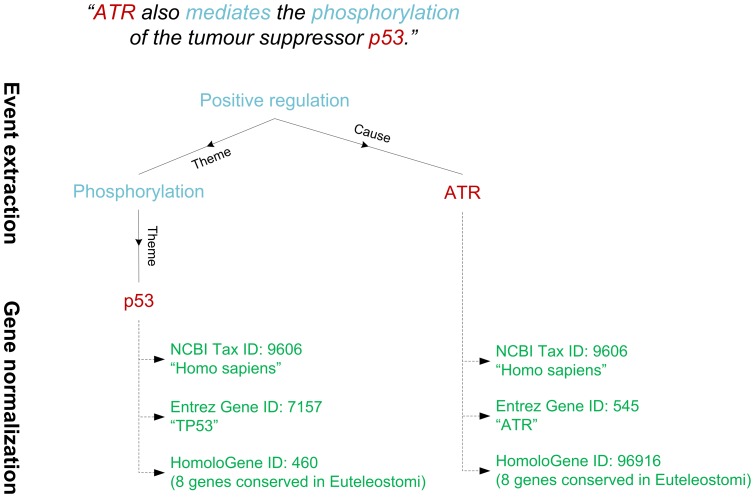
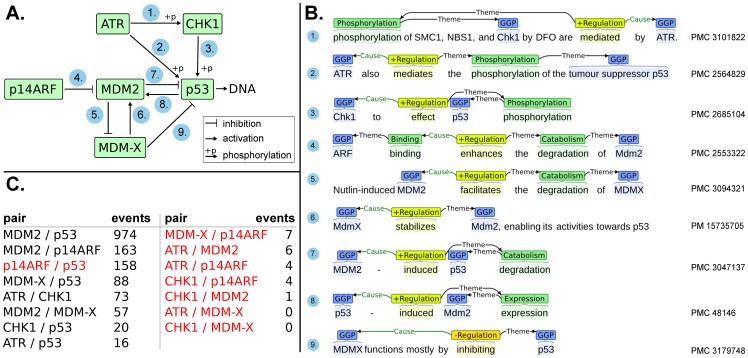
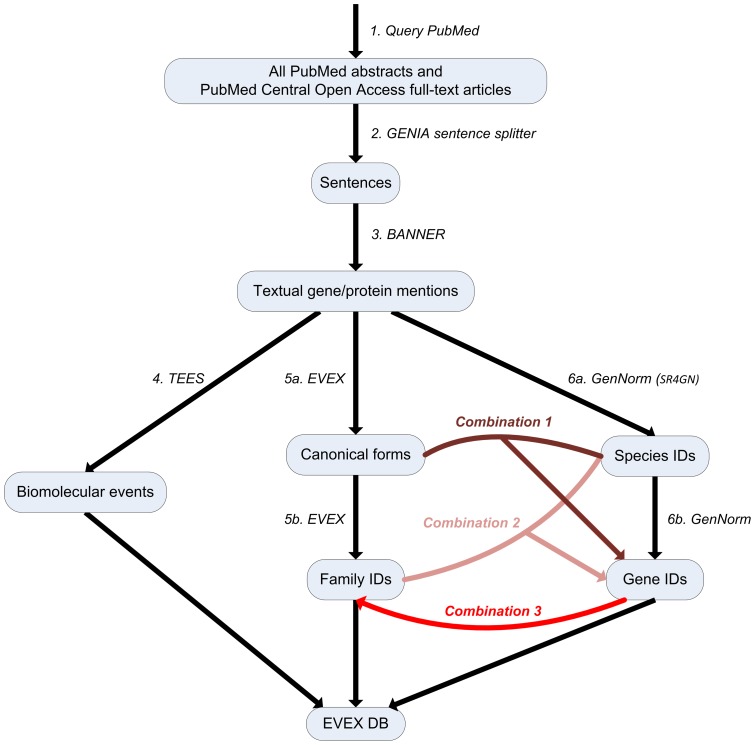
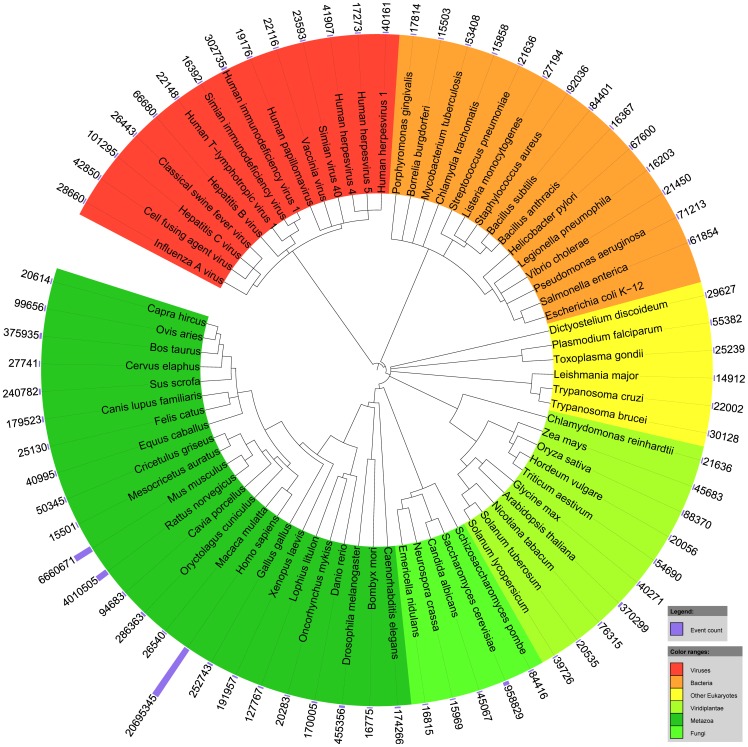
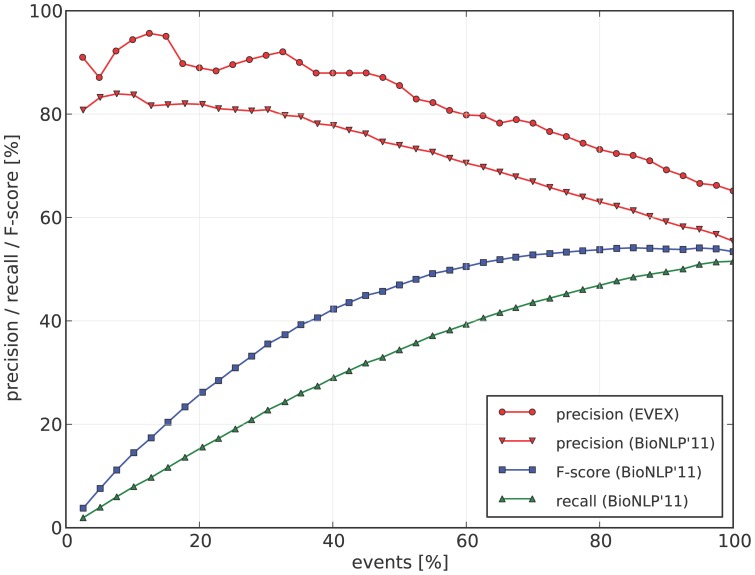
Text mining for the life sciences aims to aid database curation, knowledge summarization and information retrieval through the automated processing of biomedical texts. To provide comprehensive coverage and enable full integration with existing biomolecular database records, it is crucial that text mining tools scale up to millions of articles and that their analyses can be unambiguously linked to information recorded in resources such as UniProt, KEGG, BioGRID and NCBI databases. In this study, we investigate how fully automated text mining of complex biomolecular events can be augmented with a normalization strategy that identifies biological concepts in text, mapping them to identifiers at varying levels of granularity, ranging from canonicalized symbols to unique gene and proteins and broad gene families. To this end, we have combined two state-of-the-art text mining components, previously evaluated on two community-wide challenges, and have extended and improved upon these methods by exploiting their complementary nature. Using these systems, we perform normalization and event extraction to create a large-scale resource that is publicly available, unique in semantic scope, and covers all 21.9 million PubMed abstracts and 460 thousand PubMed Central open access full-text articles. This dataset contains 40 million biomolecular events involving 76 million gene/protein mentions, linked to 122 thousand distinct genes from 5032 species across the full taxonomic tree. Detailed evaluations and analyses reveal promising results for application of this data in database and pathway curation efforts. The main software components used in this study are released under an open-source license. Further, the resulting dataset is freely accessible through a novel API, providing programmatic and customized access (http://www.evexdb.org/api/v001/). Finally, to allow for large-scale bioinformatic analyses, the entire resource is available for bulk download from http://evexdb.org/download/, under the Creative Commons - Attribution - Share Alike (CC BY-SA) license.
文本挖掘旨在通过自动处理生物医学文本来辅助数据库管理、知识总结和信息检索。为了提供全面的覆盖范围并能够与 UniProt、KEGG、BioGRID 和 NCBI 等资源中记录的信息完全集成,文本挖掘工具必须能够扩展到数百万篇文章,并且其分析可以明确链接到这些资源中的信息。在这项研究中,我们研究了如何通过一种标准化策略来增强对复杂生物分子事件的完全自动化文本挖掘,该策略可以识别文本中的生物概念,并将它们映射到不同粒度级别的标识符,从规范符号到唯一基因和蛋白质以及广泛的基因家族。为此,我们结合了两个最先进的文本挖掘组件,这两个组件之前在两个社区范围内的挑战中进行了评估,并通过利用它们的互补性对这些方法进行了扩展和改进。使用这些系统,我们执行标准化和事件提取,创建一个大规模资源,该资源是公开的、语义范围独特的,涵盖了所有 2190 万篇 PubMed 摘要和 46 万篇 PubMed Central 开放获取全文文章。这个数据集包含 4 亿个涉及 7600 万个基因/蛋白质提及的生物分子事件,链接到来自 5032 个物种的 122000 个不同基因,这些物种跨越整个分类树。详细的评估和分析显示,该数据在数据库和途径管理工作中的应用具有很大的潜力。本研究中使用的主要软件组件是在开源许可证下发布的。此外,通过一个新的 API 可以免费访问生成的数据集,提供编程和定制访问(http://www.evexdb.org/api/v001/)。最后,为了允许进行大规模的生物信息学分析,整个资源可从 http://evexdb.org/download/ 批量下载,采用的是知识共享署名-相同方式共享(CC BY-SA)许可。