Department of Biology, Duke University, Durham, North Carolina 27708, USA.
Genetics. 2013 Jul;194(3):753-67. doi: 10.1534/genetics.113.151217. Epub 2013 May 1.
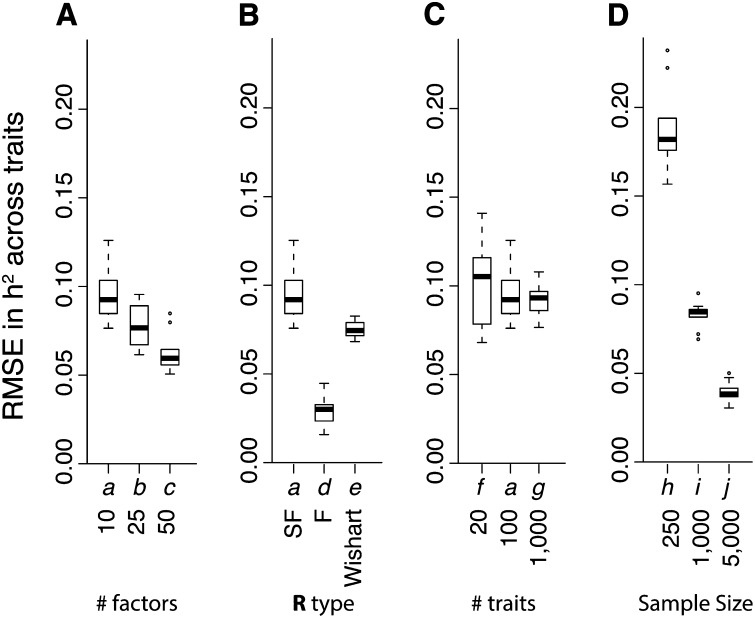
Quantitative genetic studies that model complex, multivariate phenotypes are important for both evolutionary prediction and artificial selection. For example, changes in gene expression can provide insight into developmental and physiological mechanisms that link genotype and phenotype. However, classical analytical techniques are poorly suited to quantitative genetic studies of gene expression where the number of traits assayed per individual can reach many thousand. Here, we derive a Bayesian genetic sparse factor model for estimating the genetic covariance matrix (G-matrix) of high-dimensional traits, such as gene expression, in a mixed-effects model. The key idea of our model is that we need consider only G-matrices that are biologically plausible. An organism's entire phenotype is the result of processes that are modular and have limited complexity. This implies that the G-matrix will be highly structured. In particular, we assume that a limited number of intermediate traits (or factors, e.g., variations in development or physiology) control the variation in the high-dimensional phenotype, and that each of these intermediate traits is sparse - affecting only a few observed traits. The advantages of this approach are twofold. First, sparse factors are interpretable and provide biological insight into mechanisms underlying the genetic architecture. Second, enforcing sparsity helps prevent sampling errors from swamping out the true signal in high-dimensional data. We demonstrate the advantages of our model on simulated data and in an analysis of a published Drosophila melanogaster gene expression data set.
定量遗传学研究对进化预测和人工选择都很重要,因为它可以对复杂的多变量表型进行建模。例如,基因表达的变化可以深入了解基因型和表型之间的发育和生理机制。然而,经典的分析技术在对基因表达进行定量遗传研究时效果不佳,因为每个个体所测量的性状数量可能达到数千个。在这里,我们推导出了一个贝叶斯遗传稀疏因子模型,用于在混合效应模型中估计高维性状(如基因表达)的遗传协方差矩阵(G-矩阵)。我们模型的关键思想是,我们只需要考虑在生物学上合理的 G-矩阵。一个生物体的整个表型是由模块化和有限复杂性的过程产生的。这意味着 G-矩阵将具有高度的结构性。具体来说,我们假设只有有限数量的中间性状(或因子,例如发育或生理学上的变化)控制着高维表型的变化,并且每个中间性状都是稀疏的——只影响少数观察到的性状。这种方法有两个优点。首先,稀疏因子具有可解释性,并为遗传结构的基础机制提供了生物学见解。其次,强制稀疏有助于防止采样误差淹没高维数据中的真实信号。我们在模拟数据和对已发表的黑腹果蝇基因表达数据集的分析中展示了我们模型的优势。