Department of Microbiology, Immunology and Molecular Genetics, University of California Los Angeles, Los Angeles, California, United States of America.
PLoS Pathog. 2013;9(4):e1003297. doi: 10.1371/journal.ppat.1003297. Epub 2013 Apr 18.
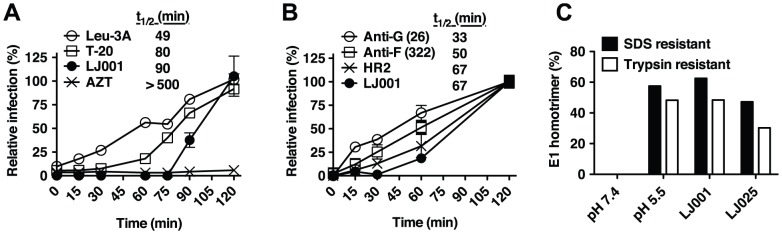
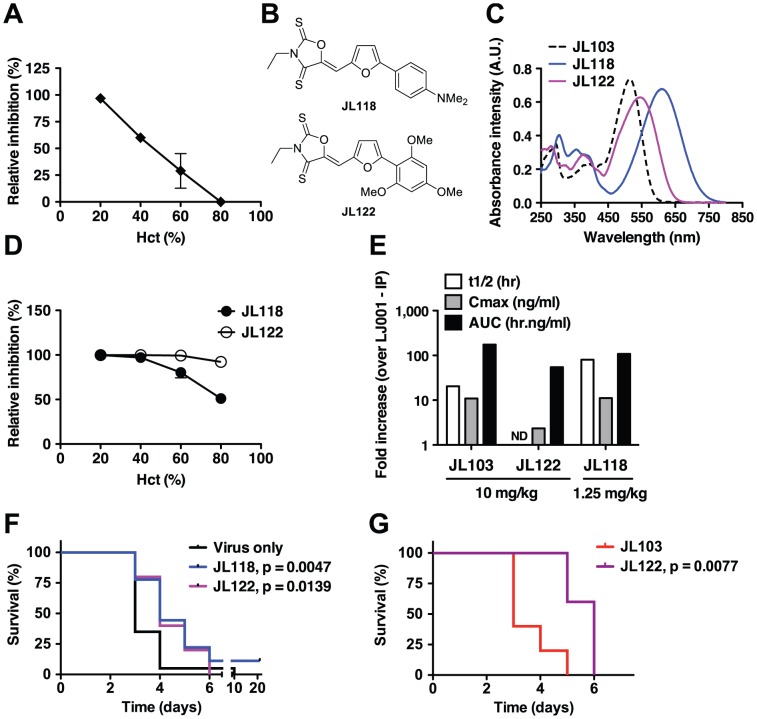
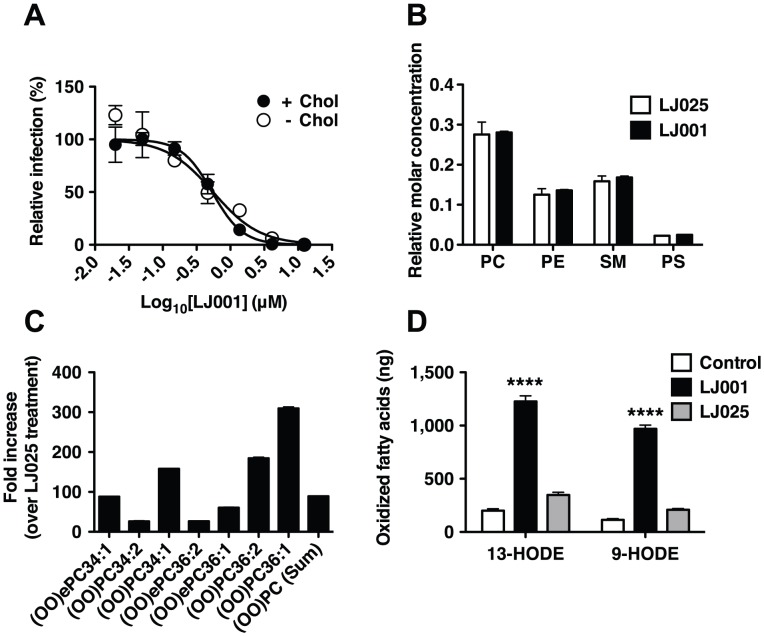
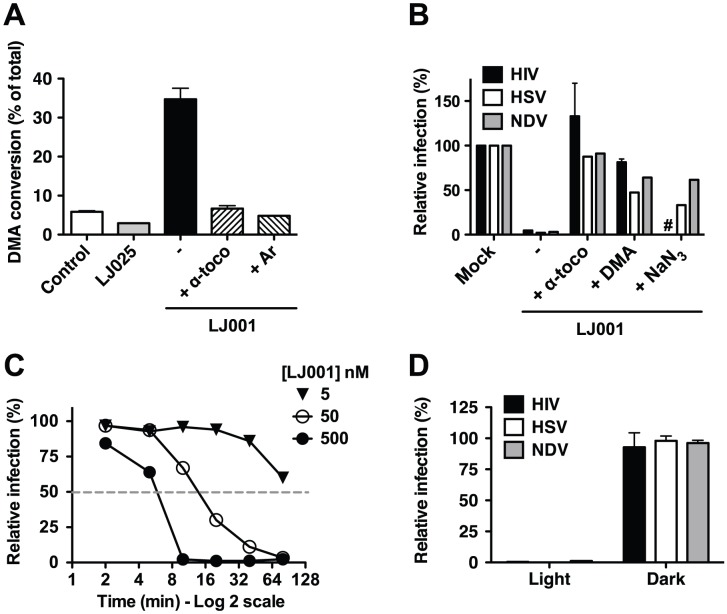
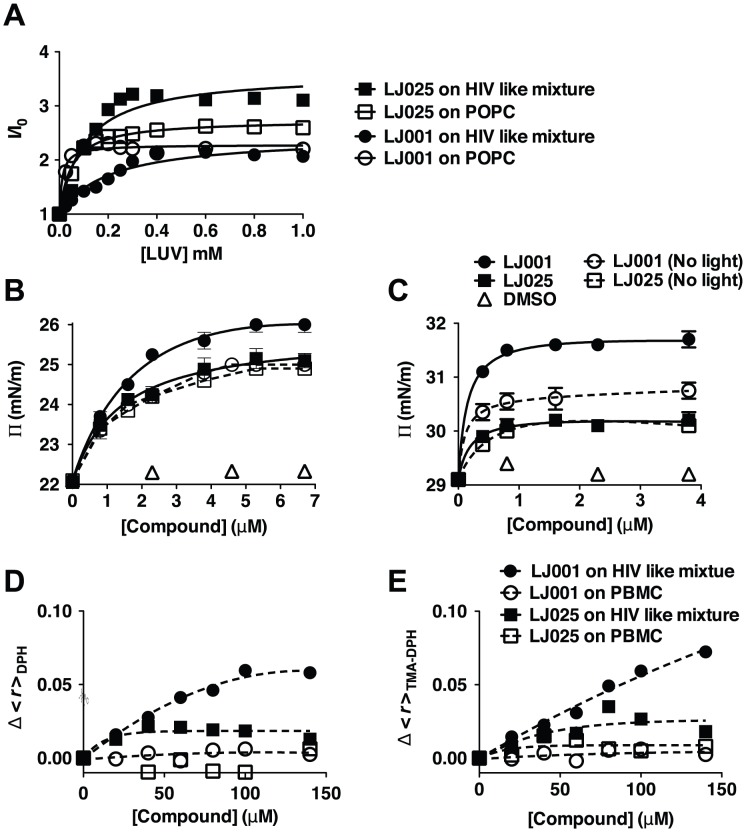
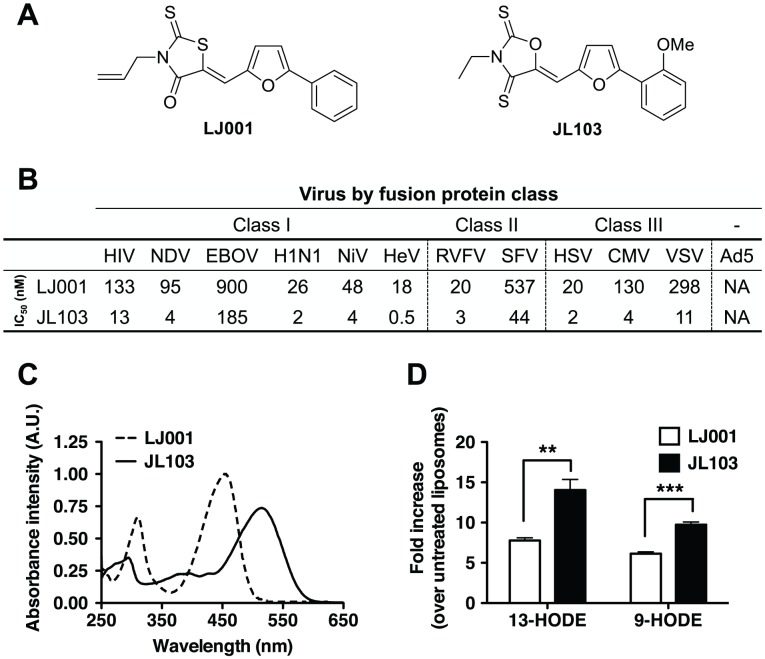
LJ001 is a lipophilic thiazolidine derivative that inhibits the entry of numerous enveloped viruses at non-cytotoxic concentrations (IC50 ≤ 0.5 µM), and was posited to exploit the physiological difference between static viral membranes and biogenic cellular membranes. We now report on the molecular mechanism that results in LJ001's specific inhibition of virus-cell fusion. The antiviral activity of LJ001 was light-dependent, required the presence of molecular oxygen, and was reversed by singlet oxygen ((1)O2) quenchers, qualifying LJ001 as a type II photosensitizer. Unsaturated phospholipids were the main target modified by LJ001-generated (1)O2. Hydroxylated fatty acid species were detected in model and viral membranes treated with LJ001, but not its inactive molecular analog, LJ025. (1)O2-mediated allylic hydroxylation of unsaturated phospholipids leads to a trans-isomerization of the double bond and concurrent formation of a hydroxyl group in the middle of the hydrophobic lipid bilayer. LJ001-induced (1)O2-mediated lipid oxidation negatively impacts on the biophysical properties of viral membranes (membrane curvature and fluidity) critical for productive virus-cell membrane fusion. LJ001 did not mediate any apparent damage on biogenic cellular membranes, likely due to multiple endogenous cytoprotection mechanisms against phospholipid hydroperoxides. Based on our understanding of LJ001's mechanism of action, we designed a new class of membrane-intercalating photosensitizers to overcome LJ001's limitations for use as an in vivo antiviral agent. Structure activity relationship (SAR) studies led to a novel class of compounds (oxazolidine-2,4-dithiones) with (1) 100-fold improved in vitro potency (IC50<10 nM), (2) red-shifted absorption spectra (for better tissue penetration), (3) increased quantum yield (efficiency of (1)O2 generation), and (4) 10-100-fold improved bioavailability. Candidate compounds in our new series moderately but significantly (p≤0.01) delayed the time to death in a murine lethal challenge model of Rift Valley Fever Virus (RVFV). The viral membrane may be a viable target for broad-spectrum antivirals that target virus-cell fusion.
LJ001 是一种脂溶性噻唑烷衍生物,可在非细胞毒性浓度下(IC50≤0.5µM)抑制多种包膜病毒的进入,据推测它利用了静态病毒膜和生物源细胞膜之间的生理差异。我们现在报告导致 LJ001 特异性抑制病毒-细胞融合的分子机制。LJ001 的抗病毒活性依赖于光,需要分子氧的存在,并且可以被单线态氧((1)O2)淬灭剂逆转,这使 LJ001 成为 II 型光敏剂。不饱和磷脂是 LJ001 产生的 (1)O2 主要修饰的靶标。在 LJ001 处理的模型和病毒膜中检测到羟基脂肪酸物质,但在其无活性的分子类似物 LJ025 中未检测到。(1)O2 介导的不饱和磷脂的烯丙基羟化导致双键的反式异构体化,并在疏水性脂质双层的中间同时形成羟基。LJ001 诱导的 (1)O2 介导的脂质氧化对病毒膜的生物物理特性(膜曲率和流动性)产生负面影响,这些特性对有效的病毒-细胞膜融合至关重要。LJ001 不会对生物源细胞膜造成任何明显的损伤,这可能是由于存在多种针对磷脂氢过氧化物的内源性细胞保护机制。基于我们对 LJ001 作用机制的理解,我们设计了一类新的膜插入型光敏剂,以克服 LJ001 作为体内抗病毒药物的局限性。构效关系(SAR)研究导致了一类新型化合物(恶唑烷-2,4-二硫酮)的出现,其具有(1)100 倍提高的体外效力(IC50<10 nM),(2)红移的吸收光谱(更好的组织穿透性),(3)增加的量子产率((1)O2 生成效率),以及(4)10-100 倍提高的生物利用度。我们新系列中的候选化合物在裂谷热病毒(RVFV)的小鼠致死性挑战模型中适度但显著(p≤0.01)延迟了死亡时间。病毒膜可能是一种可行的广谱抗病毒药物靶点,针对病毒-细胞融合。