Latif Haythem, Lerman Joshua A, Portnoy Vasiliy A, Tarasova Yekaterina, Nagarajan Harish, Schrimpe-Rutledge Alexandra C, Smith Richard D, Adkins Joshua N, Lee Dae-Hee, Qiu Yu, Zengler Karsten
Department of Bioengineering, University of California San Diego, La Jolla, California, United States of America.
PLoS Genet. 2013 Apr;9(4):e1003485. doi: 10.1371/journal.pgen.1003485. Epub 2013 Apr 25.
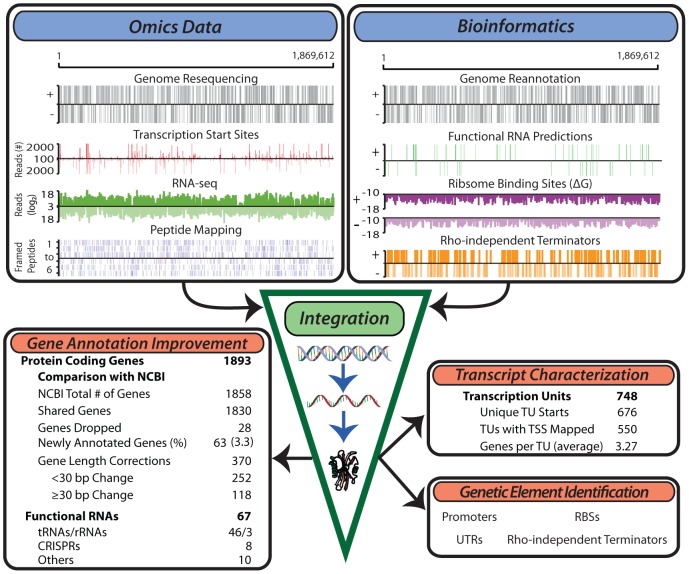
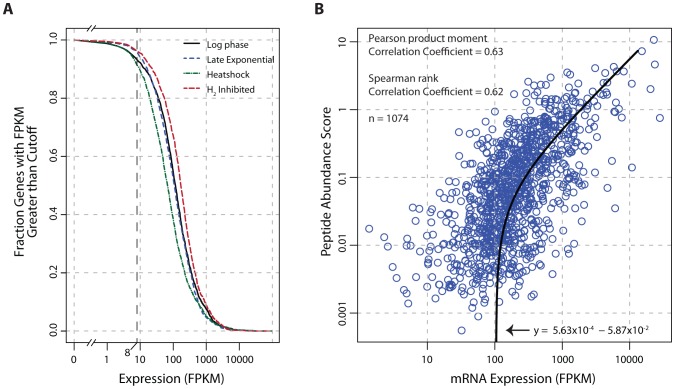
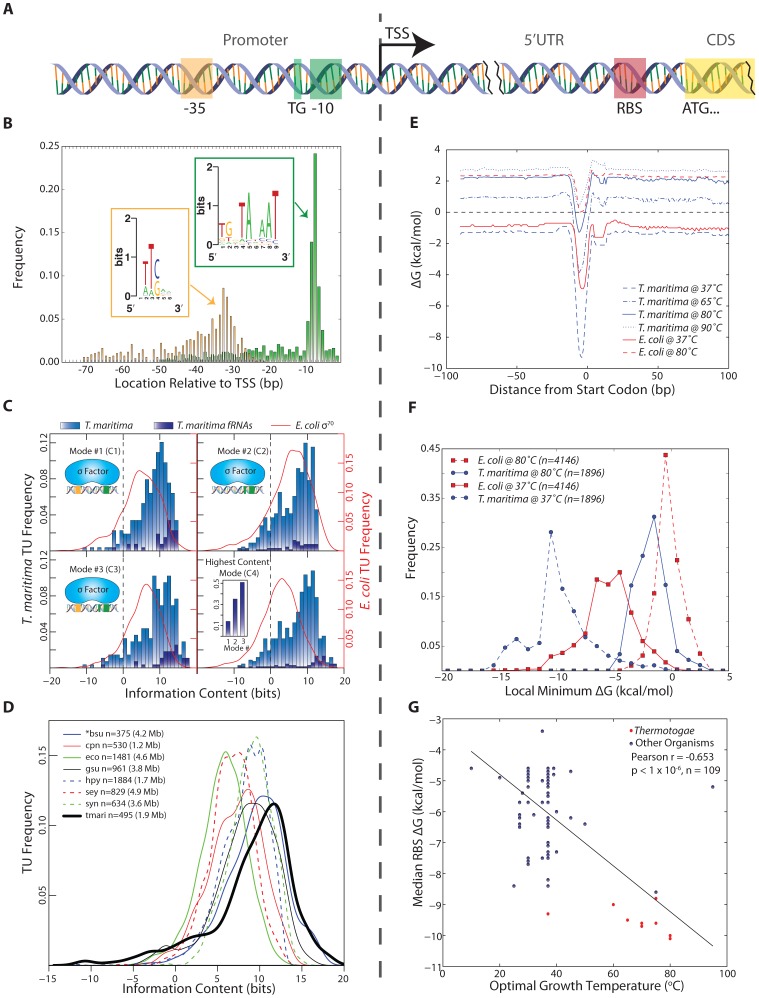
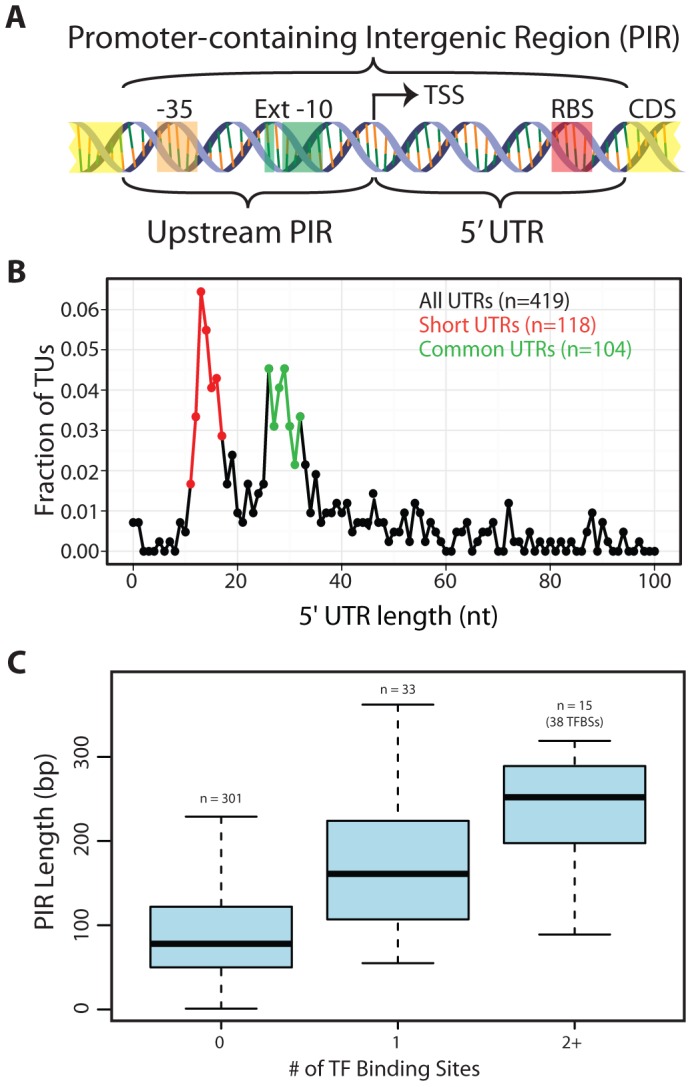
The generation of genome-scale data is becoming more routine, yet the subsequent analysis of omics data remains a significant challenge. Here, an approach that integrates multiple omics datasets with bioinformatics tools was developed that produces a detailed annotation of several microbial genomic features. This methodology was used to characterize the genome of Thermotoga maritima--a phylogenetically deep-branching, hyperthermophilic bacterium. Experimental data were generated for whole-genome resequencing, transcription start site (TSS) determination, transcriptome profiling, and proteome profiling. These datasets, analyzed in combination with bioinformatics tools, served as a basis for the improvement of gene annotation, the elucidation of transcription units (TUs), the identification of putative non-coding RNAs (ncRNAs), and the determination of promoters and ribosome binding sites. This revealed many distinctive properties of the T. maritima genome organization relative to other bacteria. This genome has a high number of genes per TU (3.3), a paucity of putative ncRNAs (12), and few TUs with multiple TSSs (3.7%). Quantitative analysis of promoters and ribosome binding sites showed increased sequence conservation relative to other bacteria. The 5'UTRs follow an atypical bimodal length distribution comprised of "Short" 5'UTRs (11-17 nt) and "Common" 5'UTRs (26-32 nt). Transcriptional regulation is limited by a lack of intergenic space for the majority of TUs. Lastly, a high fraction of annotated genes are expressed independent of growth state and a linear correlation of mRNA/protein is observed (Pearson r = 0.63, p<2.2 × 10(-16) t-test). These distinctive properties are hypothesized to be a reflection of this organism's hyperthermophilic lifestyle and could yield novel insights into the evolutionary trajectory of microbial life on earth.
基因组规模数据的生成正变得越来越常规化,然而对组学数据的后续分析仍然是一项重大挑战。在此,开发了一种将多个组学数据集与生物信息学工具相结合的方法,该方法可对多种微生物基因组特征进行详细注释。此方法被用于表征嗜热栖热菌(Thermotoga maritima)的基因组——一种系统发育上处于深分支的嗜热细菌。生成了用于全基因组重测序、转录起始位点(TSS)测定、转录组分析和蛋白质组分析的实验数据。这些数据集与生物信息学工具结合进行分析,为改进基因注释、阐明转录单元(TU)、鉴定假定的非编码RNA(ncRNA)以及确定启动子和核糖体结合位点提供了基础。这揭示了嗜热栖热菌基因组组织相对于其他细菌的许多独特特性。该基因组每个TU的基因数量较多(3.3个),假定的ncRNA较少(12个),具有多个TSS的TU也较少(3.7%)。对启动子和核糖体结合位点的定量分析表明,相对于其他细菌,其序列保守性有所增加。5'非翻译区(UTR)呈现出一种非典型的双峰长度分布,由“短”5'UTR(11 - 17个核苷酸)和“常见”5'UTR(26 - 32个核苷酸)组成。大多数TU缺乏基因间空间,限制了转录调控。最后,很大一部分注释基因的表达与生长状态无关,并且观察到mRNA/蛋白质之间存在线性相关性(Pearson相关系数r = 0.63,t检验p < 2.2 × 10(-16))。据推测,这些独特特性反映了这种生物的嗜热生活方式,并且可能为地球上微生物生命的进化轨迹提供新的见解。